Transcriptome Analysis of the Procession from Chronic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Methylglyoxal Down-Regulates the Expression of Cell Cycle Associated
www.nature.com/scientificreports OPEN Methylglyoxal down-regulates the expression of cell cycle associated genes and activates the p53 Received: 8 May 2018 Accepted: 12 December 2018 pathway in human umbilical vein Published: xx xx xxxx endothelial cells Jana D. Braun1, Diego O. Pastene1, Annette Breedijk1, Angelica Rodriguez1, Björn B. Hofmann1, Carsten Sticht2, Elke von Ochsenstein3, Heike Allgayer3, Jacob van den Born4, Stephan Bakker4, Sibylle J. Hauske1, Bernhard K. Krämer1, Benito A. Yard1 & Thomas Albrecht1 Although methylglyoxal (MGO) has emerged as key mediator of diabetic microvascular complications, the infuence of MGO on the vascular transcriptome has not thoroughly been assessed. Since diabetes is associated with low grade infammation causing sustained nuclear factor-kappa B (NF-κB) activation, the current study addressed 1) to what extent MGO changes the transcriptome of human umbilical vein endothelial cells (HUVECs) exposed to an infammatory milieu, 2) what are the dominant pathways by which these changes occur and 3) to what extent is this afected by carnosine, a putative scavenger of MGO. Microarray analysis revealed that exposure of HUVECs to high MGO concentrations signifcantly changes gene expression, characterized by prominent down-regulation of cell cycle associated genes and up-regulation of heme oxygenase-1 (HO-1). KEGG-based pathway analysis identifed six signifcantly enriched pathways of which the p53 pathway was the most afected. No signifcant enrichment of infammatory pathways was found, yet, MGO did inhibit VCAM-1 expression in Western blot analysis. Carnosine signifcantly counteracted MGO-mediated changes in a subset of diferentially expressed genes. Collectively, our results suggest that MGO initiates distinct transcriptional changes in cell cycle/apoptosis genes, which may explain MGO toxicity at high concentrations. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptNat Immunol Author Manuscript. Author manuscript; Author Manuscript available in PMC 2009 May 01. Published in final edited form as: Nat Immunol. 2008 November ; 9(11): 1307–1315. doi:10.1038/ni.1662. The actin regulator coronin-1A is mutated in a thymic egress deficient mouse strain and in a T−B+NK+ SCID patient Lawrence R. Shiow1,2,3, David W. Roadcap5, Kenneth Paris7, Susan R. Watson2, Irina L. Grigorova1,2, Tonya Lebet2,4, Jinping An1,2, Ying Xu1,2, Craig N. Jenne1,2, Niko Föger8, Ricardo U. Sorensen7, Christopher C. Goodnow6, James E. Bear5, Jennifer M. Puck3,4, and Jason G. Cyster1,2,3 1 Howard Hughes Medical Institute, University of California San Francisco, San Francisco, CA 2 Department of Microbiology and Immunology, University of California San Francisco, San Francisco, CA 3 Biomedical Sciences Graduate Program, University of California San Francisco, San Francisco, CA 4 Department of Pediatrics and Institute for Human Genetics, University of California San Francisco, San Francisco, CA 5 Lineberger Comprehensive Cancer Center and Department of Cell and Developmental Biology, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 6 John Curtin School of Medical Research, The Australian National University, Canberra, Australia 7 Department of Pediatrics, Louisiana State University Health Sciences Center and Children’s Hospital, New Orleans, LA 8 Department of Immunology and Cell Biology, Leibniz Center for Medicine and Biosciences, Borstel, Germany Abstract Mice carrying the recessive peripheral T cell deficiency (Ptcd) locus have a block in thymic egress but the mechanism responsible is undefined. -
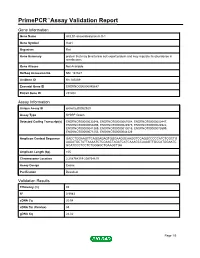
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name HCLS1-associated protein X-1 Gene Symbol Hax1 Organism Rat Gene Summary protein that may bind to bile salt export protein and may regulate its abundance in membranes Gene Aliases Not Available RefSeq Accession No. NM_181627 UniGene ID Rn.185269 Ensembl Gene ID ENSRNOG00000045647 Entrez Gene ID 291202 Assay Information Unique Assay ID qRnoCED0052920 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000030348, ENSRNOT00000067004, ENSRNOT00000000447, ENSRNOT00000059496, ENSRNOT00000024975, ENSRNOT00000024922, ENSRNOT00000041389, ENSRNOT00000010015, ENSRNOT00000073599, ENSRNOT00000071253, ENSRNOT00000044326 Amplicon Context Sequence GACCTGGAAGTTCAGGAGAGTGGGAAGGCAAGGTCCAGGCCCCCATCTCGCTG AAGATGCTATTAAAATCTCGAACTAGATCATCAAAGCCAAAGTTGCCATGGAATC GCATCCCTCCTCTGGGGCTGAAGCTGA Amplicon Length (bp) 105 Chromosome Location 2:208764319-208764619 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 92 R2 0.9983 cDNA Cq 20.54 cDNA Tm (Celsius) 83 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report Hax1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Identification and Validation of Genes Affecting Aortic Lesions in Mice
Identification and validation of genes affecting aortic lesions in mice Xia Yang, … , Aldons J. Lusis, Pek Yee Lum J Clin Invest. 2010;120(7):2414-2422. https://doi.org/10.1172/JCI42742. Research Article Cardiology Atherosclerosis represents the most significant risk factor for coronary artery disease (CAD), the leading cause of death in developed countries. To better understand the pathogenesis of atherosclerosis, we applied a likelih ood-based model selection method to infer gene-disease causality relationships for the aortic lesion trait in a segregating mouse population demonstrating a spectrum of susceptibility to developing atherosclerotic lesions. We identified 292 genes that tested causal for aortic lesions from liver and adipose tissues of these mice, and we experimentally validated one of these candidate causal genes, complement component 3a receptor 1 (C3ar1), using a knockout mouse model. We also found that genes identified by this method overlapped with genes progressively regulated in the aortic arches of 2 mouse models of atherosclerosis during atherosclerotic lesion development. By comparing our gene set with findings from public human genome-wide association studies (GWAS) of CAD and related traits, we found that 5 genes identified by our study overlapped with published studies in humans in which they were identified as risk factors for multiple atherosclerosis- related pathologies, including myocardial infarction, serum uric acid levels, mean platelet volume, aortic root size, and heart failure. Candidate causal genes were also found to be enriched with CAD risk polymorphisms identified by the Wellcome Trust Case Control Consortium (WTCCC). Our findings therefore validate the ability of causality testing procedures to […] Find the latest version: https://jci.me/42742/pdf Research article Identification and validation of genes affecting aortic lesions in mice Xia Yang,1,2 Larry Peterson,3 Rolf Thieringer,4 Joshua L. -

Identification of the Fatty Acid Synthase Interaction Network Via Itraq-Based Proteomics Indicates the Potential Molecular Mecha
Huang et al. Cancer Cell Int (2020) 20:332 https://doi.org/10.1186/s12935-020-01409-2 Cancer Cell International PRIMARY RESEARCH Open Access Identifcation of the fatty acid synthase interaction network via iTRAQ-based proteomics indicates the potential molecular mechanisms of liver cancer metastasis Juan Huang1, Yao Tang1, Xiaoqin Zou1, Yi Lu1, Sha She1, Wenyue Zhang1, Hong Ren1, Yixuan Yang1,2* and Huaidong Hu1,2* Abstract Background: Fatty acid synthase (FASN) is highly expressed in various types of cancer and has an important role in carcinogenesis and metastasis. To clarify the mechanisms of FASN in liver cancer invasion and metastasis, the FASN protein interaction network in liver cancer was identifed by targeted proteomic analysis. Methods: Wound healing and Transwell assays was performed to observe the efect of FASN during migration and invasion in liver cancer. Isobaric tags for relative and absolute quantitation (iTRAQ)-based mass spectrometry were used to identify proteins interacting with FASN in HepG2 cells. Diferential expressed proteins were validated by co-immunoprecipitation, western blot analyses and confocal microscopy. Western blot and reverse transcription- quantitative polymerase chain reaction (RT-qPCR) were performed to demonstrate the mechanism of FASN regulating metastasis. Results: FASN knockdown inhibited migration and invasion of HepG2 and SMMC7721 cells. A total of, 79 proteins interacting with FASN were identifed. Additionally, gene ontology term enrichment analysis indicated that the majority of biological regulation and cellular processes that the FASN-interacting proteins were associated with. Co- precipitation and co-localization of FASN with fascin actin-bundling protein 1 (FSCN1), signal-induced proliferation- associated 1 (SIPA1), spectrin β, non-erythrocytic 1 (SPTBN1) and CD59 were evaluated. -

Prediction of Candidate Primary Immunodeficiency Disease Genes
DNA RESEARCH 16, 345–351, (2009) doi:10.1093/dnares/dsp019 Prediction of Candidate Primary Immunodeficiency Disease Genes Using a Support Vector Machine Learning Approach SHIVAKUMAR Keerthikumar1,2,3,SAHELY Bhadra4,KUMARAN Kandasamy1,2,5,RAJESH Raju1,2,3, Y.L. Ramachandra2,CHIRANJIB Bhattacharyya4,KOHSUKE Imai8,OSAMU Ohara6,7, SUJATHA Mohan1,3, and AKHILESH Pandey1,5,* Institute of Bioinformatics, International Technology Park, Bangalore 560 066, India1; Department of Biotechnology and Bioinformatics, Kuvempu University, Jnanasahyadri, Shimoga 577 451, India2; Research Unit for Immunoinformatics, Research Center for Allergy and Immunology, RIKEN Yokohama Institute, Kanagawa 230- 0045, Japan3; Department of Computer Science and Automation, Indian Institute of Science, Bangalore 560 012, India4; McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, 733 N. Broadway, BRB Room 527, Baltimore, MD 21205, USA5; Laboratory for Immunogenomics, Research Center for Allergy and Immunology, RIKEN, Yokohama Institute, Kanagawa 230-0045, Japan6; Department of Human Genome Technology, Kazusa DNA Research Institute, 2-6-7 Kazusa-Kamatari, Kisarazu, Chiba 292-0818, Japan7 and Department of Medical Informatics, National Defense Medical College, Saitama 359-8513, Japan8 (Received 23 July 2009; accepted 5 September 2009; published online 3 October 2009) Abstract Screening and early identification of primary immunodeficiency disease (PID) genes is a major chal- lenge for physicians. Many resources have catalogued molecular alterations in known PID genes along with their associated clinical and immunological phenotypes. However, these resources do not assist in identifying candidate PID genes. We have recently developed a platform designated Resource of Asian PDIs, which hosts information pertaining to molecular alterations, protein–protein interaction networks, mouse studies and microarray gene expression profiling of all known PID genes. -

Associated Fibroblasts in Non-Small Cell Lung Cancer
Prognostic gene-expression signature of carcinoma- associated fibroblasts in non-small cell lung cancer Roya Navaba,1, Dan Strumpfa,1, Bizhan Bandarchia,1, Chang-Qi Zhua,1, Melania Pintiliea, Varune Rohan Ramnarinea, Emin Ibrahimova, Nikolina Radulovicha, Lisa Leunga, Malgorzata Barczyka,b, Devang Panchala, Christine Toa, James J. Yuna, Sandy Dera, Frances A. Shepherda,c, Igor Jurisicaa,d,e, and Ming-Sound Tsaoa,e,f,2 aThe Campbell Family Institute for Cancer Research, Ontario Cancer Institute at Princess Margaret Hospital, University Health Network, Toronto, ON, Canada M5G 2M9; bDepartment of Biomedicine, Jonas Lies vei, N-5009 Bergen, Norway; Departments of cMedicine, dComputer Science, and eMedical Biophysics, and fLaboratory of Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada M5A 2N4 Edited* by Tak Wah Mak, The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute at Princess Margaret Hospital, University Health Network, Toronto, ON, Canada, and approved March 10, 2011 (received for review September 28, 2010) The tumor microenvironment strongly influences cancer develop- Results ment, progression, and metastasis. The role of carcinoma-associated Cultured CAFs Display Features of Myofibroblasts. By using a study fibroblasts (CAFs) in these processes and their clinical impact has protocol approved by the Institutional Research Ethics Board, not been studied systematically in non-small cell lung carcinoma CAFs and NFs were cultured from 15 surgically resected primary (NSCLC). We established primary cultures of CAFs and matched nor- NSCLCs, and the histologically confirmed normal lung tissue was mal fibroblasts (NFs) from 15 resected NSCLC. We demonstrate that obtained from the same lobe (Table S1A). Both the primary CAFs have greater ability than NFs to enhance the tumorigenicity of cultured CAFs and tumor stromal fibroblasts expressed α-smooth lung cancer cell lines. -

WNT16 Is a New Marker of Senescence
Table S1. A. Complete list of 177 genes overexpressed in replicative senescence Value Gene Description UniGene RefSeq 2.440 WNT16 wingless-type MMTV integration site family, member 16 (WNT16), transcript variant 2, mRNA. Hs.272375 NM_016087 2.355 MMP10 matrix metallopeptidase 10 (stromelysin 2) (MMP10), mRNA. Hs.2258 NM_002425 2.344 MMP3 matrix metallopeptidase 3 (stromelysin 1, progelatinase) (MMP3), mRNA. Hs.375129 NM_002422 2.300 HIST1H2AC Histone cluster 1, H2ac Hs.484950 2.134 CLDN1 claudin 1 (CLDN1), mRNA. Hs.439060 NM_021101 2.119 TSPAN13 tetraspanin 13 (TSPAN13), mRNA. Hs.364544 NM_014399 2.112 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.070 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.026 DCBLD2 discoidin, CUB and LCCL domain containing 2 (DCBLD2), mRNA. Hs.203691 NM_080927 2.007 SERPINB2 serpin peptidase inhibitor, clade B (ovalbumin), member 2 (SERPINB2), mRNA. Hs.594481 NM_002575 2.004 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.989 OBFC2A Oligonucleotide/oligosaccharide-binding fold containing 2A Hs.591610 1.962 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.947 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 2, mRNA. Hs.472101 NM_182797 1.934 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 1, mRNA. Hs.472101 NM_000933 1.933 KRTAP1-5 keratin associated protein 1-5 (KRTAP1-5), mRNA. Hs.534499 NM_031957 1.894 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.884 CYTL1 cytokine-like 1 (CYTL1), mRNA. Hs.13872 NM_018659 tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (TNFRSF10D), 1.848 TNFRSF10D Hs.213467 NM_003840 mRNA.