A Crash Course in Statistical Mechanics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Path Integrals in Quantum Mechanics
Path Integrals in Quantum Mechanics Dennis V. Perepelitsa MIT Department of Physics 70 Amherst Ave. Cambridge, MA 02142 Abstract We present the path integral formulation of quantum mechanics and demon- strate its equivalence to the Schr¨odinger picture. We apply the method to the free particle and quantum harmonic oscillator, investigate the Euclidean path integral, and discuss other applications. 1 Introduction A fundamental question in quantum mechanics is how does the state of a particle evolve with time? That is, the determination the time-evolution ψ(t) of some initial | i state ψ(t ) . Quantum mechanics is fully predictive [3] in the sense that initial | 0 i conditions and knowledge of the potential occupied by the particle is enough to fully specify the state of the particle for all future times.1 In the early twentieth century, Erwin Schr¨odinger derived an equation specifies how the instantaneous change in the wavefunction d ψ(t) depends on the system dt | i inhabited by the state in the form of the Hamiltonian. In this formulation, the eigenstates of the Hamiltonian play an important role, since their time-evolution is easy to calculate (i.e. they are stationary). A well-established method of solution, after the entire eigenspectrum of Hˆ is known, is to decompose the initial state into this eigenbasis, apply time evolution to each and then reassemble the eigenstates. That is, 1In the analysis below, we consider only the position of a particle, and not any other quantum property such as spin. 2 D.V. Perepelitsa n=∞ ψ(t) = exp [ iE t/~] n ψ(t ) n (1) | i − n h | 0 i| i n=0 X This (Hamiltonian) formulation works in many cases. -

Key Concepts for Future QIS Learners Workshop Output Published Online May 13, 2020
Key Concepts for Future QIS Learners Workshop output published online May 13, 2020 Background and Overview On behalf of the Interagency Working Group on Workforce, Industry and Infrastructure, under the NSTC Subcommittee on Quantum Information Science (QIS), the National Science Foundation invited 25 researchers and educators to come together to deliberate on defining a core set of key concepts for future QIS learners that could provide a starting point for further curricular and educator development activities. The deliberative group included university and industry researchers, secondary school and college educators, and representatives from educational and professional organizations. The workshop participants focused on identifying concepts that could, with additional supporting resources, help prepare secondary school students to engage with QIS and provide possible pathways for broader public engagement. This workshop report identifies a set of nine Key Concepts. Each Concept is introduced with a concise overall statement, followed by a few important fundamentals. Connections to current and future technologies are included, providing relevance and context. The first Key Concept defines the field as a whole. Concepts 2-6 introduce ideas that are necessary for building an understanding of quantum information science and its applications. Concepts 7-9 provide short explanations of critical areas of study within QIS: quantum computing, quantum communication and quantum sensing. The Key Concepts are not intended to be an introductory guide to quantum information science, but rather provide a framework for future expansion and adaptation for students at different levels in computer science, mathematics, physics, and chemistry courses. As such, it is expected that educators and other community stakeholders may not yet have a working knowledge of content covered in the Key Concepts. -

Classical Mechanics
Classical Mechanics Hyoungsoon Choi Spring, 2014 Contents 1 Introduction4 1.1 Kinematics and Kinetics . .5 1.2 Kinematics: Watching Wallace and Gromit ............6 1.3 Inertia and Inertial Frame . .8 2 Newton's Laws of Motion 10 2.1 The First Law: The Law of Inertia . 10 2.2 The Second Law: The Equation of Motion . 11 2.3 The Third Law: The Law of Action and Reaction . 12 3 Laws of Conservation 14 3.1 Conservation of Momentum . 14 3.2 Conservation of Angular Momentum . 15 3.3 Conservation of Energy . 17 3.3.1 Kinetic energy . 17 3.3.2 Potential energy . 18 3.3.3 Mechanical energy conservation . 19 4 Solving Equation of Motions 20 4.1 Force-Free Motion . 21 4.2 Constant Force Motion . 22 4.2.1 Constant force motion in one dimension . 22 4.2.2 Constant force motion in two dimensions . 23 4.3 Varying Force Motion . 25 4.3.1 Drag force . 25 4.3.2 Harmonic oscillator . 29 5 Lagrangian Mechanics 30 5.1 Configuration Space . 30 5.2 Lagrangian Equations of Motion . 32 5.3 Generalized Coordinates . 34 5.4 Lagrangian Mechanics . 36 5.5 D'Alembert's Principle . 37 5.6 Conjugate Variables . 39 1 CONTENTS 2 6 Hamiltonian Mechanics 40 6.1 Legendre Transformation: From Lagrangian to Hamiltonian . 40 6.2 Hamilton's Equations . 41 6.3 Configuration Space and Phase Space . 43 6.4 Hamiltonian and Energy . 45 7 Central Force Motion 47 7.1 Conservation Laws in Central Force Field . 47 7.2 The Path Equation . -
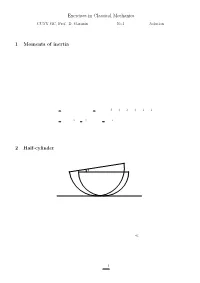
Exercises in Classical Mechanics 1 Moments of Inertia 2 Half-Cylinder
Exercises in Classical Mechanics CUNY GC, Prof. D. Garanin No.1 Solution ||||||||||||||||||||||||||||||||||||||||| 1 Moments of inertia (5 points) Calculate tensors of inertia with respect to the principal axes of the following bodies: a) Hollow sphere of mass M and radius R: b) Cone of the height h and radius of the base R; both with respect to the apex and to the center of mass. c) Body of a box shape with sides a; b; and c: Solution: a) By symmetry for the sphere I®¯ = I±®¯: (1) One can ¯nd I easily noticing that for the hollow sphere is 1 1 X ³ ´ I = (I + I + I ) = m y2 + z2 + z2 + x2 + x2 + y2 3 xx yy zz 3 i i i i i i i i 2 X 2 X 2 = m r2 = R2 m = MR2: (2) 3 i i 3 i 3 i i b) and c): Standard solutions 2 Half-cylinder ϕ (10 points) Consider a half-cylinder of mass M and radius R on a horizontal plane. a) Find the position of its center of mass (CM) and the moment of inertia with respect to CM. b) Write down the Lagrange function in terms of the angle ' (see Fig.) c) Find the frequency of cylinder's oscillations in the linear regime, ' ¿ 1. Solution: (a) First we ¯nd the distance a between the CM and the geometrical center of the cylinder. With σ being the density for the cross-sectional surface, so that ¼R2 M = σ ; (3) 2 1 one obtains Z Z 2 2σ R p σ R q a = dx x R2 ¡ x2 = dy R2 ¡ y M 0 M 0 ¯ 2 ³ ´3=2¯R σ 2 2 ¯ σ 2 3 4 = ¡ R ¡ y ¯ = R = R: (4) M 3 0 M 3 3¼ The moment of inertia of the half-cylinder with respect to the geometrical center is the same as that of the cylinder, 1 I0 = MR2: (5) 2 The moment of inertia with respect to the CM I can be found from the relation I0 = I + Ma2 (6) that yields " µ ¶ # " # 1 4 2 1 32 I = I0 ¡ Ma2 = MR2 ¡ = MR2 1 ¡ ' 0:3199MR2: (7) 2 3¼ 2 (3¼)2 (b) The Lagrange function L is given by L('; '_ ) = T ('; '_ ) ¡ U('): (8) The potential energy U of the half-cylinder is due to the elevation of its CM resulting from the deviation of ' from zero. -
![Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals](https://docslib.b-cdn.net/cover/4942/arxiv-1910-10745v1-cond-mat-str-el-23-oct-2019-2-2-symmetry-protected-time-crystals-304942.webp)
Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals
A Brief History of Time Crystals Vedika Khemania,b,∗, Roderich Moessnerc, S. L. Sondhid aDepartment of Physics, Harvard University, Cambridge, Massachusetts 02138, USA bDepartment of Physics, Stanford University, Stanford, California 94305, USA cMax-Planck-Institut f¨urPhysik komplexer Systeme, 01187 Dresden, Germany dDepartment of Physics, Princeton University, Princeton, New Jersey 08544, USA Abstract The idea of breaking time-translation symmetry has fascinated humanity at least since ancient proposals of the per- petuum mobile. Unlike the breaking of other symmetries, such as spatial translation in a crystal or spin rotation in a magnet, time translation symmetry breaking (TTSB) has been tantalisingly elusive. We review this history up to recent developments which have shown that discrete TTSB does takes place in periodically driven (Floquet) systems in the presence of many-body localization (MBL). Such Floquet time-crystals represent a new paradigm in quantum statistical mechanics — that of an intrinsically out-of-equilibrium many-body phase of matter with no equilibrium counterpart. We include a compendium of the necessary background on the statistical mechanics of phase structure in many- body systems, before specializing to a detailed discussion of the nature, and diagnostics, of TTSB. In particular, we provide precise definitions that formalize the notion of a time-crystal as a stable, macroscopic, conservative clock — explaining both the need for a many-body system in the infinite volume limit, and for a lack of net energy absorption or dissipation. Our discussion emphasizes that TTSB in a time-crystal is accompanied by the breaking of a spatial symmetry — so that time-crystals exhibit a novel form of spatiotemporal order. -

On Thermality of CFT Eigenstates
On thermality of CFT eigenstates Pallab Basu1 , Diptarka Das2 , Shouvik Datta3 and Sridip Pal2 1International Center for Theoretical Sciences-TIFR, Shivakote, Hesaraghatta Hobli, Bengaluru North 560089, India. 2Department of Physics, University of California San Diego, La Jolla, CA 92093, USA. 3Institut f¨urTheoretische Physik, Eidgen¨ossischeTechnische Hochschule Z¨urich, 8093 Z¨urich,Switzerland. E-mail: [email protected], [email protected], [email protected], [email protected] Abstract: The Eigenstate Thermalization Hypothesis (ETH) provides a way to under- stand how an isolated quantum mechanical system can be approximated by a thermal density matrix. We find a class of operators in (1+1)-d conformal field theories, consisting of quasi-primaries of the identity module, which satisfy the hypothesis only at the leading order in large central charge. In the context of subsystem ETH, this plays a role in the deviation of the reduced density matrices, corresponding to a finite energy density eigen- state from its hypothesized thermal approximation. The universal deviation in terms of the square of the trace-square distance goes as the 8th power of the subsystem fraction and is suppressed by powers of inverse central charge (c). Furthermore, the non-universal deviations from subsystem ETH are found to be proportional to the heavy-light-heavy p arXiv:1705.03001v2 [hep-th] 26 Aug 2017 structure constants which are typically exponentially suppressed in h=c, where h is the conformal scaling dimension of the finite energy density -

From Quantum Chaos and Eigenstate Thermalization to Statistical
Advances in Physics,2016 Vol. 65, No. 3, 239–362, http://dx.doi.org/10.1080/00018732.2016.1198134 REVIEW ARTICLE From quantum chaos and eigenstate thermalization to statistical mechanics and thermodynamics a,b c b a Luca D’Alessio ,YarivKafri,AnatoliPolkovnikov and Marcos Rigol ∗ aDepartment of Physics, The Pennsylvania State University, University Park, PA 16802, USA; bDepartment of Physics, Boston University, Boston, MA 02215, USA; cDepartment of Physics, Technion, Haifa 32000, Israel (Received 22 September 2015; accepted 2 June 2016) This review gives a pedagogical introduction to the eigenstate thermalization hypothesis (ETH), its basis, and its implications to statistical mechanics and thermodynamics. In the first part, ETH is introduced as a natural extension of ideas from quantum chaos and ran- dom matrix theory (RMT). To this end, we present a brief overview of classical and quantum chaos, as well as RMT and some of its most important predictions. The latter include the statistics of energy levels, eigenstate components, and matrix elements of observables. Build- ing on these, we introduce the ETH and show that it allows one to describe thermalization in isolated chaotic systems without invoking the notion of an external bath. We examine numerical evidence of eigenstate thermalization from studies of many-body lattice systems. We also introduce the concept of a quench as a means of taking isolated systems out of equi- librium, and discuss results of numerical experiments on quantum quenches. The second part of the review explores the implications of quantum chaos and ETH to thermodynamics. Basic thermodynamic relations are derived, including the second law of thermodynamics, the fun- damental thermodynamic relation, fluctuation theorems, the fluctuation–dissipation relation, and the Einstein and Onsager relations. -

Lecture 2: Basics of Quantum Mechanics
Lecture 2: Basics of Quantum Mechanics Scribed by: Vitaly Feldman Department of Mathematics, MIT September 9, 2003 In this lecture we will cover the basics of Quantum Mechanics which are required to under stand the process of quantum computation. To simplify the discussion we assume that all the Hilbert spaces mentioned below are finitedimensional. The process of quantum computation can be abstracted via the following four postulates. Postulate 1. Associated to any isolated physical system is a complex vector space with inner product (that is, a Hilbert space) known as the state space of the system. The state of the system is completely described by a unit vector in this space. Qubits were the example of such system that we saw in the previous lecture. In its physical realization as a polarization of a photon we have two basis vectors: |�� and |↔� representing the vertical and the horizontal polarizations respectively. In this basis vector polarized at angle θ can be expressed as cos θ |↔� − sin θ |��. An important property of a quantum system is that multiplying a quantum state by a unit complex factor (eiθ ) yields the same complex state. Therefore eiθ |�� and |�� represent essentially the same state. Notation 1. State χ is denoted by |χ� (often called a ket) is a column vector, e.g., ⎛ ⎞ √1/2 ⎝ i 3/2 ⎠ 0 † |χ� = �χ| (often √called a bra) denotes a conjugate transpose of |χ�. In the previous example we would get (1/2, −i 3/2, 0). It is easy to verify that �χ| χ� = 1 and �x|y� ≤ 1. -

The Development of the Action Principle a Didactic History from Euler-Lagrange to Schwinger Series: Springerbriefs in Physics
springer.com Walter Dittrich The Development of the Action Principle A Didactic History from Euler-Lagrange to Schwinger Series: SpringerBriefs in Physics Provides a philosophical interpretation of the development of physics Contains many worked examples Shows the path of the action principle - from Euler's first contributions to present topics This book describes the historical development of the principle of stationary action from the 17th to the 20th centuries. Reference is made to the most important contributors to this topic, in particular Bernoullis, Leibniz, Euler, Lagrange and Laplace. The leading theme is how the action principle is applied to problems in classical physics such as hydrodynamics, electrodynamics and gravity, extending also to the modern formulation of quantum mechanics 1st ed. 2021, XV, 135 p. 23 illus., 1 illus. and quantum field theory, especially quantum electrodynamics. A critical analysis of operator in color. versus c-number field theory is given. The book contains many worked examples. In particular, the term "vacuum" is scrutinized. The book is aimed primarily at actively working researchers, Printed book graduate students and historians interested in the philosophical interpretation and evolution of Softcover physics; in particular, in understanding the action principle and its application to a wide range 54,99 € | £49.99 | $69.99 of natural phenomena. [1]58,84 € (D) | 60,49 € (A) | CHF 65,00 eBook 46,00 € | £39.99 | $54.99 [2]46,00 € (D) | 46,00 € (A) | CHF 52,00 Available from your library or springer.com/shop MyCopy [3] Printed eBook for just € | $ 24.99 springer.com/mycopy Order online at springer.com / or for the Americas call (toll free) 1-800-SPRINGER / or email us at: [email protected]. -

Quantum Mechanics
Quantum Mechanics Richard Fitzpatrick Professor of Physics The University of Texas at Austin Contents 1 Introduction 5 1.1 Intendedaudience................................ 5 1.2 MajorSources .................................. 5 1.3 AimofCourse .................................. 6 1.4 OutlineofCourse ................................ 6 2 Probability Theory 7 2.1 Introduction ................................... 7 2.2 WhatisProbability?.............................. 7 2.3 CombiningProbabilities. ... 7 2.4 Mean,Variance,andStandardDeviation . ..... 9 2.5 ContinuousProbabilityDistributions. ........ 11 3 Wave-Particle Duality 13 3.1 Introduction ................................... 13 3.2 Wavefunctions.................................. 13 3.3 PlaneWaves ................................... 14 3.4 RepresentationofWavesviaComplexFunctions . ....... 15 3.5 ClassicalLightWaves ............................. 18 3.6 PhotoelectricEffect ............................. 19 3.7 QuantumTheoryofLight. .. .. .. .. .. .. .. .. .. .. .. .. .. 21 3.8 ClassicalInterferenceofLightWaves . ...... 21 3.9 QuantumInterferenceofLight . 22 3.10 ClassicalParticles . .. .. .. .. .. .. .. .. .. .. .. .. .. .. 25 3.11 QuantumParticles............................... 25 3.12 WavePackets .................................. 26 2 QUANTUM MECHANICS 3.13 EvolutionofWavePackets . 29 3.14 Heisenberg’sUncertaintyPrinciple . ........ 32 3.15 Schr¨odinger’sEquation . 35 3.16 CollapseoftheWaveFunction . 36 4 Fundamentals of Quantum Mechanics 39 4.1 Introduction .................................. -

Outline of Physical Science
Outline of physical science “Physical Science” redirects here. It is not to be confused • Astronomy – study of celestial objects (such as stars, with Physics. galaxies, planets, moons, asteroids, comets and neb- ulae), the physics, chemistry, and evolution of such Physical science is a branch of natural science that stud- objects, and phenomena that originate outside the atmosphere of Earth, including supernovae explo- ies non-living systems, in contrast to life science. It in turn has many branches, each referred to as a “physical sions, gamma ray bursts, and cosmic microwave background radiation. science”, together called the “physical sciences”. How- ever, the term “physical” creates an unintended, some- • Branches of astronomy what arbitrary distinction, since many branches of physi- cal science also study biological phenomena and branches • Chemistry – studies the composition, structure, of chemistry such as organic chemistry. properties and change of matter.[8][9] In this realm, chemistry deals with such topics as the properties of individual atoms, the manner in which atoms form 1 What is physical science? chemical bonds in the formation of compounds, the interactions of substances through intermolecular forces to give matter its general properties, and the Physical science can be described as all of the following: interactions between substances through chemical reactions to form different substances. • A branch of science (a systematic enterprise that builds and organizes knowledge in the form of • Branches of chemistry testable explanations and predictions about the • universe).[1][2][3] Earth science – all-embracing term referring to the fields of science dealing with planet Earth. Earth • A branch of natural science – natural science science is the study of how the natural environ- is a major branch of science that tries to ex- ment (ecosphere or Earth system) works and how it plain and predict nature’s phenomena, based evolved to its current state. -

Articles and Thus Only Consider the Mo- with Solar Radiation
Hydrol. Earth Syst. Sci., 17, 225–251, 2013 www.hydrol-earth-syst-sci.net/17/225/2013/ Hydrology and doi:10.5194/hess-17-225-2013 Earth System © Author(s) 2013. CC Attribution 3.0 License. Sciences Thermodynamics, maximum power, and the dynamics of preferential river flow structures at the continental scale A. Kleidon1, E. Zehe2, U. Ehret2, and U. Scherer2 1Max-Planck Institute for Biogeochemistry, Hans-Knoll-Str.¨ 10, 07745 Jena, Germany 2Institute of Water Resources and River Basin Management, Karlsruhe Institute of Technology – KIT, Karlsruhe, Germany Correspondence to: A. Kleidon ([email protected]) Received: 14 May 2012 – Published in Hydrol. Earth Syst. Sci. Discuss.: 11 June 2012 Revised: 14 December 2012 – Accepted: 22 December 2012 – Published: 22 January 2013 Abstract. The organization of drainage basins shows some not randomly diffuse through the soil to the ocean, but rather reproducible phenomena, as exemplified by self-similar frac- collects in channels that are organized in tree-like structures tal river network structures and typical scaling laws, and along topographic gradients. This organization of surface these have been related to energetic optimization principles, runoff into tree-like structures of river networks is not a pe- such as minimization of stream power, minimum energy ex- culiar exception, but is persistent and can generally be found penditure or maximum “access”. Here we describe the or- in many different regions of the Earth. Hence, it would seem ganization and dynamics of drainage systems using thermo- that the evolution and maintenance of these structures of river dynamics, focusing on the generation, dissipation and trans- networks is a reproducible phenomenon that would be the ex- fer of free energy associated with river flow and sediment pected outcome of how natural systems organize their flows.