An Interactive Multi-View Genome Browser for Next-Generation Comparative Microorganism Genomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Apache Flex 2
flex #flex 1 1: flex 2 2 2 Examples 2 Apache Flex 2 Apache FlexApache Flex SDK 2 Apache Flex SDK 3 3 3 3 3 4 Adobe AIR SDK 4 Adobe Flash Player 4 Adobe Flash Player playerglobal.swc 5 Adobe Pixel Bender 5 5 5 Hello World 6 Hello World 6 7 You can share this PDF with anyone you feel could benefit from it, downloaded the latest version from: flex It is an unofficial and free flex ebook created for educational purposes. All the content is extracted from Stack Overflow Documentation, which is written by many hardworking individuals at Stack Overflow. It is neither affiliated with Stack Overflow nor official flex. The content is released under Creative Commons BY-SA, and the list of contributors to each chapter are provided in the credits section at the end of this book. Images may be copyright of their respective owners unless otherwise specified. All trademarks and registered trademarks are the property of their respective company owners. Use the content presented in this book at your own risk; it is not guaranteed to be correct nor accurate, please send your feedback and corrections to [email protected] https://riptutorial.com/zh-TW/home 1 1: flex Apache FlexApache Foundation。SWFFlashAdobe AirAndroidiOSBlackBerryWindowsOSX。 FlexUIMXMLActionScript3。SWF。 FlexRESTSOAPJSONJMSAMFJavaPHPRuby.NET。 Apache Flex SDKHTTPServiceWebServiceRemoteObject。 Flex。 Apache FlexApache License v.2.0 。SDKSDK。 WebJava.NET。 Apache FlexApacheApache Software Foundation。 Apache Flex SDK 4.15 2016111 Apache Flex SDK 4.14.1 2015331 Apache Flex SDK 4.14.0 201523 Apache Flex SDK 4.13.0 2014728 Apache Flex SDK 4.12.1 201453 Apache Flex SDK 4.12.0 2014310 Apache Flex SDK 4.11.0 20131028 Apache Flex SDK 4.10.0 201386 Apache Flex SDK 4.9.0 2013228 Examples Apache Flex Apache Flex SDK。Apache Flex SDKAdobe AIRWindowsOS X / macOS。。 Apache FlexApache Flex SDK https://riptutorial.com/zh-TW/home 2 1. -

Leveraging Declarative Languages in Web Application Development
World Wide Web DOI 10.1007/s11280-015-0339-z Leveraging declarative languages in web application development Petri Vuorimaa & Markku Laine & Evgenia Litvinova & Denis Shestakov Received: 7 February 2014 /Revised: 24 February 2015 /Accepted: 4 March 2015 # The Author(s) 2015. This article is published with open access at Springerlink.com Abstract Web Applications have become an omnipresent part of our daily lives. They are easy to use, but hard to develop. WYSIWYG editors, form builders, mashup editors, and markup authoring tools ease the development of Web Applications. However, more advanced Web Applications require servers-side programming, which is beyond the skills of end-user developers. In this paper, we discuss how declarative languages can simplify Web Application development and empower end-users as Web developers. We first identify nine end-user Web Application development levels ranging from simple visual customization to advanced three- tier programming. Then, we propose expanding the presentation tier to support all aspects of Web Application development. We introduce a unified XForms-based framework—called XFormsDB—that supports both client-side and server-side Web Application development. Furthermore, we make a language extension proposal—called XFormsRTC—for adding true real-time communication capabilities to XForms. We also present XFormsDB Integrated Development Environment (XIDE), which assists end-users in authoring highly interactive data-driven Web Applications. XIDE supports all Web Application development levels and, especially, promotes the transition from markup authoring and snippet programming to single and unified language programming. Keywords Web framework . Web application . Web development . End-user development . Declarative language . Real-time communication P. Vuorimaa (*) : M. -

Silk Test 17.5
Silk Test 17.5 Silk4J User Guide Micro Focus The Lawn 22-30 Old Bath Road Newbury, Berkshire RG14 1QN UK http://www.microfocus.com Copyright © Micro Focus 2016. All rights reserved. MICRO FOCUS, the Micro Focus logo and Silk Test are trademarks or registered trademarks of Micro Focus IP Development Limited or its subsidiaries or affiliated companies in the United States, United Kingdom and other countries. All other marks are the property of their respective owners. 2016-11-03 ii Contents Welcome to Silk4J 17.5 ......................................................................................9 Licensing Information ......................................................................................10 Silk4J ................................................................................................................ 11 Do I Need Administrator Privileges to Run Silk4J? ........................................................... 11 Best Practices for Using Silk4J ........................................................................................ 11 Automation Under Special Conditions (Missing Peripherals) ............................................12 Silk Test Product Suite ...................................................................................................... 13 What's New in Silk4J ........................................................................................15 Mobile Device Live View ................................................................................................... 15 Testing Hybrid -

Multi-Project Management
Multi-Project Management DATA SHEET Multi-Project Management It is a challenge to monitor and control a large number of projects within a company or division. It is even more challenging the more the projects influence each other or have mutual dependencies and compete for the same resources. The tasks in multi-project management essentially include: . Analyzing target compliance (scope, costs, deadlines) . Analyzing the status and progress of projects . Identifying critical plan deviations to allow early corrective action to be taken . Consistent quality management and consistent project evaluation . Standardizing project processes (e.g. formalized proposals, phase models for particular project types, etc.) and monitoring compliance . Creating aggregated reports and decision support for management . Comprehensive knowledge management (project learning and implementation into new pro jects) . Documenting decisions © OPUS Business Solutions GmbH 2018 Page 1/7 Multi-Project Management The KLUSA Management module To carry out the tasks described above, KLUSA is organized according to typical tasks and procedures. In order to handle a large number of projects, there are filters to limit and select the projects displayed. All reports and evaluations then relate to the filtered project list. Filters can be individually saved by users, and important filters can be made available to all users centrally from the project office. In addition, the KLUSA portals can be applied to limit the scope of project analysis. Example of a project phase overview in KLUSA Status and progress of projects The status overviews offered by KLUSA include the following: . Dashboard . Phases and project deadlines (planned and actual deadlines) . Summary . Milestone overview . Costs and benefits (planned, actual, forecast costs) . -

Omi Troubleshooting Guide
Operations Manager i Software Version: 10.63 Troubleshooting Document Release Date: December 2017 Software Release Date: November 2017 Troubleshooting Legal Notices Warranty The only warranties for Seattle SpinCo, Inc and its subsidiaries (“Seattle”) products and services are set forth in the express warranty statements accompanying such products and services. Nothing herein should be construed as constituting an additional warranty. Seattle shall not be liable for technical or editorial errors or omissions contained herein. The information contained herein is subject to change without notice. Restricted Rights Legend Confidential computer software. Except as specifically indicated, valid license from Seattle required for possession, use or copying. Consistent with FAR 12.211 and 12.212, Commercial Computer Software, Computer Software Documentation, and Technical Data for Commercial Items are licensed to the U.S. Government under vendor's standard commercial license. Copyright Notice © 2015 - 2017 EntIT Software LLC, a Micro Focus company Trademark Notices Adobe® and Acrobat® are trademarks of Adobe Systems Incorporated. AMD, the AMD Arrow symbol and ATI are trademarks of Advanced Micro Devices, Inc. Citrix® and XenDesktop® are registered trademarks of Citrix Systems, Inc. and/or one more of its subsidiaries, and may be registered in the United States Patent and Trademark Office and in other countries. Google™ and Google Maps™ are trademarks of Google Inc. Intel®, Itanium®, Pentium®, and Intel® Xeon® are trademarks of Intel Corporation in the U.S. and other countries. iPad® and iPhone® are trademarks of Apple Inc. Java is a registered trademark of Oracle and/or its affiliates. Linux® is the registered trademark of Linus Torvalds in the U.S. -
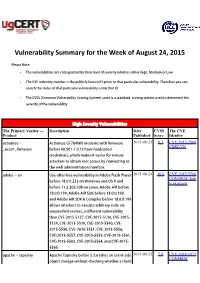
Vulnerability Summary for the Week of August 24, 2015
Vulnerability Summary for the Week of August 24, 2015 Please Note: • The vulnerabilities are cattegorized by their level of severity which is either High, Medium or Low. • The !" indentity number is the #ublicly $nown %& given to that #articular vulnerability. Therefore you can search the status of that #articular vulnerability using that %&. • The !'S (Common !ulnerability 'coring System) score is a standard scoring system used to determine the severity of the vulnerability. High Severity Vulnerabilities The Primary Vendor --- Description Date CVSS The CVE Product Published Score Identity actiontec ** .ctiontec /T0123N modems with firmware 2015-08-23 8.3 CVE-2015-2904 CERT-VN +ncs,-+firmware before NC',-*-.0.14 have hardcoded credentials, which ma$es it easier for remote attac$ers to obtain root access by connecting to the web administration interface. adobe ** air 5se*after*free vulnerability in .dobe 6lash Player 2015-08-24 10.0 CVE-2015-5566 CONFIRM (link before -1.0.0.747 on 3indows and 8' 9 and is external) before --.2.2,7.5,1 on Linu;, .dobe .%< before -1.0.,.1==, .dobe .%< '&> before -1.0.0.-==, and .dobe .%< '&> ? om#iler before -1.0.0.1== allows attac$ers to e;ecute arbitrary code via uns#ecified vectors, a different vulnerability than !"*7,-:*:-70, !"*7,-:*:-4,, !"*7,-:* :-42, !"*7,-:*::4=, !"*7,-:*::2,, !"* 7,-:*:::,, !"*7,-:*:::-, !"*7,-:*:::@, !"*7,-:*:::0, !"*7,-:*:::=, !"*7,-:*::@-, !"*7,-:*::@4, !"*7,-:*::@2, and !"*7,-:* ::@:. a#ache ** ta#estry .#ache Ta#estry before :.3.6 relies on client*side 2015-08-22 7.8 CVE-2014-1972 CONFIRM obAect storage without chec$ing whether a client has modified an obAect, which allows remote CONFIRM JVNDB (link is attac$ers to cause a denial of service (resource external) consum#tion) or e;ecute arbitrary code via JVN (link is crafted serialized data. -

Upkit: an Open-Source, Portable, and Lightweight Update Framework For
Paper published at the 39th IEEE International Conference on Distributed Computing Systems (ICDCS) in July 2019 UpKit: An Open-Source, Portable, and Lightweight Update Framework for Constrained IoT Devices Antonio Langiu, Carlo Alberto Boano, Markus Schuß, and Kay Romer¨ Institute of Technical Informatics, Graz University of Technology, Austria E-mail: [email protected]; [email protected]; [email protected]; [email protected] Abstract—Updating the software running on constrained IoT to embed some software update capabilities in the firmware devices such as low-power sensors and actuators in a secure and running on each IoT device [6], [7]. Such capabilities include, efficient way is an open problem. The limited computational, among others, the (over-the-air) download of the update image, memory, and storage capabilities of these devices, together with their small energy budget, indeed, restrict the number of features the verification of its integrity and authenticity, as well as its that can be embedded into an update system and make it installation [8]. All these steps should be performed while also difficult to build a generic and compact solution. As a minimizing the downtime of a device and its services. result, existing update systems for constrained IoT devices are often not portable, do not perform a proper verification of the Updating constrained IoT devices. Updating the software downloaded firmware, or focus only on a single phase of the running on constrained IoT devices, e.g., low-power sensors update process, which exposes them to security threats and calls and actuators [9], is still an open research problem [6], for new solutions. -

Arcgis and Flex Web Applications Kevin Deege @Kdeege
Esri International User Conference San Diego, California Technical Workshops | ArcGIS and Flex Web Applications Kevin Deege @kdeege Agenda • Overview • ArcGIS Application Builder • ArcGIS API for Flex • Customizing the ArcGIS Viewer for Flex • Road ahead • Additional resources What is your role? Build, configure, develop Configure Build Develop Flex Web Application What does an application need? • Content OR - Portal Web Maps - ArcGIS Online Web Maps Basemaps and - ArcGIS Online for Organizations Operational - Portal for ArcGIS Layers - Basemaps and operational Layers Layer Your Server Map ArcGISOnline Layer What does an application need? • Tools and Functionality - Components / Widgets - Tasks / API Functionality What does an application need? • Appearance - Standard - Custom Requirements – Build and Configure Build, configure • Application Builder Build - Adobe AIR • Flash Player (testing) Configure • Web server Requirements - Developer Developer configuration • Flex SDK (4.6 or higher) • Flash Builder (4.6 or higher) Develop - Adobe AIR • Flash Player (11.1 or higher) - Debugger version • ArcGIS Viewer for Flex - Source available on Github • ArcGIS API for Flex - Available on ArcGIS Resource Center Flash Runtimes and Flex SDK • Adobe - Adobe Flash Player - Adobe Flash Builder - Donated Flex SDK to Apache - Adobe Flex SDK 4.6.0 • Apache Foundation - Apache Flex 4.9.0 SDK - http://flex.apache.org/ Getting started with the Viewer - Builder Navigate to ArcGIS Resources site http://links.esri.com/flexviewer Link to download ArcGIS Viewer -

Veritas Infoscale™ 7.4 Third-Party Software License Agreements - AIX, Linux, Solaris, Windows
Veritas InfoScale™ 7.4 Third-Party Software License Agreements - AIX, Linux, Solaris, Windows Last updated: updated: 2018- 052018-31 -05-31 Veritas Technologies LLC—Confidential Veritas InfoScale™ 7.4 Third-Party Software License Agreements Table of Contents ADOBE FLEX SOFTWARE DEVELOPMENT KIT V4.0 SOFTWARE LICENSE......................................................................... 4 APACHE LICENSE 2.0 ..................................................................................................................................................... 9 ARTISTIC LICENSE 1.0 .................................................................................................................................................. 13 ARTISTIC LICENSE 1.0 (PERL) ....................................................................................................................................... 20 ARTISTIC LICENSE 2.0 .................................................................................................................................................. 24 BOOST SOFTWARE LICENSE 1.0 .................................................................................................................................. 27 BSD 2-CLAUSE "SIMPLIFIED" LICENSE ......................................................................................................................... 27 BSD 3-CLAUSE "NEW" OR "REVISED" LICENSE ............................................................................................................ 28 BSD 4-CLAUSE "ORIGINAL" -

Operations Basics
COREMEDIA CONTENT CLOUD Operations Basics Operations Basics Operations Basics | Copyright CoreMedia GmbH © 2021 CoreMedia GmbH Ludwig-Erhard-Straße 18 20459 Hamburg International All rights reserved. No part of this manual or the corresponding program may be reproduced or copied in any form (print, photocopy or other process) without the written permission of CoreMedia GmbH. Germany Alle Rechte vorbehalten. CoreMedia und weitere im Text erwähnte CoreMedia Produkte sowie die entsprechenden Logos sind Marken oder eingetragene Marken der CoreMedia GmbH in Deutschland. Alle anderen Namen von Produkten sind Marken der jeweiligen Firmen. Das Handbuch bzw. Teile hiervon sowie die dazugehörigen Programme dürfen in keiner Weise (Druck, Fotokopie oder sonstige Verfahren) ohne schriftliche Genehmigung der CoreMedia GmbH reproduziert oder vervielfältigt werden. Unberührt hiervon bleiben die gesetzlich erlaubten Nutzungsarten nach dem UrhG. Licenses and Trademarks All trademarks acknowledged. June 09, 2021 (Release 2101) COREMEDIA CONTENT CLOUD ii Operations Basics | 1. Introduction ........................................................................... 1 1.1. Audience .................................................................... 2 1.2. Typographic Conventions ................................................. 3 1.3. CoreMedia Services ........................................................ 5 1.3.1. Registration ...................................................... 5 1.3.2. CoreMedia Releases ............................................ -

María Carmen Carrascosa Baena
Next Generation of Informatics Tools for Big Data Analytics in Drug Discovery María Carmen Carrascosa Baena TESI DOCTORAL UPF / 2017 DIRECTOR DE LA TESI Dr. Jordi Mestres DEPARTAMENT CEXS ii A mi familia/ohana iii I've seen things you people wouldn't believe. Attack ships on fire off the shoulder of Orion. I watched C‐beams glitter in the dark near the Tannhäuser Gate. All those moments will be lost in time, like tears in rain. Time to die. Roy Batty Nexus 6 replicant serial code N6MAA10816 Blade Runner This is true love. You think this happen every day? William Goldman, The Princess Bride vi Summary The classical silver bullet paradigm of one drug interacting with a single target linked to a disease is currently challenged. It is now widely recognized that one drug interacts with multiple targets and these targets are involved in many biological pathways and expressed in a variety of organs. As the notion of complexity has been gradually accepted, the reductionist drug discovery approach has naturally evolved towards systems multilevel strategies. Thanks to technological advances, there has been a huge increase of data generated in the various fields relevant to drug discovery, namely, chemistry, pharmacology, toxicology, genomics, metabolomics, etc., which has expanded dramatically our ability to generate computational models with increasing performance and coverage. But ultimately, extracting knowledge from this complex, vast and heterogeneous amount of data is not straightforward. The main objective of this Thesis is to develop new interactive analytics and visualization tools and investigate their ability to extract knowledge from highly interconnected data when implemented into an integrated flexible platform to facilitate drawing simple answers from complex questions. -

Silk Test 21.0.1
Silk Test 21.0.1 Release Notes Micro Focus The Lawn 22-30 Old Bath Road Newbury, Berkshire RG14 1QN UK http://www.microfocus.com © Copyright 1992-2021 Micro Focus or one of its affiliates. MICRO FOCUS, the Micro Focus logo and Silk Test are trademarks or registered trademarks of Micro Focus or one of its affiliates. All other marks are the property of their respective owners. 2021-09-09 ii Contents Silk Test 21.0.1 Release Notes .......................................................................... 4 Silk Test Product Suite ...................................................................................... 5 System Requirements and Prerequisites ........................................................ 7 Hardware Requirements ..................................................................................................... 7 Software Requirements .......................................................................................................7 What's New in Silk Test ......................................................................................8 Image Verification with Cross-Platform Reporting ...............................................................8 Automatic Browser Support ................................................................................................ 8 Standalone Recorder .......................................................................................................... 8 AutoPass Support ..............................................................................................................