Frederickpadleyphdthesis1966
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chemical Composition of Lipids, Especially Triacylglycerol, in Grape
Agric. Biol. Chem., 54 (4), 1035-1042, 1990 1035 Chemical Composition of Lipids, Especially Triacylglycerol, in Grape Seeds Masao Ohnishi, Shuji Hirose,* Masayuki Kawaguchi,* Seisuke Ito and Yasuhiko Fujino1 Department of Agricultural Chemistry, Obihiro University of Agriculture and Veterinary Medicine, Obihiro 080, Japan * Tokachi-Ikeda Research Institute for Viticulture and Enology, Ikeda, Hokkaido 083, Japan Received November 13, 1989 Total lipids were extracted from five varieties of grape seeds and systematically analyzed for their chemical compositions. The yields of the total lipids were 10-16 %, and triacylglycerol (TG) usually amounted to c. 90 %of the whole. From a reversed-phase high-performance liquid chromatographic analysis, the major molecular species of TGwere shown to be trilinolein (40 %), oleoyldilinolein (21 %) and palmitoyldilinolein (18 %). The component fatty acids were asymmetrically distributed at C-l and C-3 of the TGmolecule. Palmitic acid was exclusively located at the C-l position, although unsaturated fatty acids, especially linoleic acid, were predominant at the C-l position, as at the C-2 and C-3 positions. Compared with TG, higher proportions of palmitic and linolenic acids were generally observed in thirteen other lipid classes isolated from grape seeds, although the fatty acid compositions of the diacylglycerol and free fatty acids were roughly identical with that of TG. As component sterols, sitosterol, campesterol and stigmasterol, especially the former, were predominant. Their relative proportions were somewhatdifferent from each other between the neutral and polar sterol lipids. In the process of wine making, large quan- Europe. It has been reported that grape seed tities of the pomace are produced as by- oils contain large amounts of unsaturated fatty products, which are utilized after fermentation acids such as linoleic and oleic acid, and mainly as a soil conditioner. -

Chemistry and Physical Properties of Estolides
GRASAS Y ACEITES, 62 (1), ENERO-MARZO, 8-20, 2011, ISSN: 0017-3495 DOI: 10.3989/gya/010810 Chemistry and physical properties of estolides By Terry A. Isbell* United States Department of Agriculture, Agriculture Research Service, National Center for Agricultural Utilization Research, 1815 N. University St. Peoria, Illinois 61529 (*Corresponding author: [email protected]) RESUMEN capped estolides of oleic that have both good low temperature properties (pour point –5 to – 39oC) and good oxidative stability. Propiedades físicas y químicas de los estólidos Estolides from meadowfoam fatty acids do not have good low temperature properties but have been extensively used in Los estólidos son una familia de compuestos sintetizados cosmetics where they provide good moisturizing properties. a partir de aceites hidroxilados como los de ricino o lesquere- lla o mediante la condensación de ácidos grasos sobre el do- KEY-WORDS: Castor – Estolide – Lesquerella – Oleic ble enlace de un segundo ácido graso insaturado. Los estóli- acid – Physical Properties – Synthesis. dos de ricino y lesquerela se derivan tanto de sus triglicéridos como de sus ácidos grasos libres empleándose el residuo hi- droxilo para formar los ésteres estólidos de los mismos. Los triglicéridos estólidos tienen puntos de fluidez crítica de entre 1. INTRODUCTION 9 y -36ºC y baja estabilidad, con tiempos de oxidación en reci- piente vacío a presión (RPVOT) de entre 29 y 52 minutos in- Estolides are natural and synthetic compounds cluso con la adición de un 1% de una mezcla antioxidante a derived from fats and oils. The estolide structure las muestras. Estas propiedades contrastan con las de los es- is identified by the secondary ester linkage of one tólidos de ácido lesquerólico y ricinoleico, que poseen puntos fatty acyl molecule to the alkyl backbone of another críticos de fluidez mucho más bajos (de -36 a -54). -
![[ Agr. Biol. Chem., Vol. 29, No. 2, P. 111-116, 1965] Glyceride Structure](https://docslib.b-cdn.net/cover/4532/agr-biol-chem-vol-29-no-2-p-111-116-1965-glyceride-structure-284532.webp)
[ Agr. Biol. Chem., Vol. 29, No. 2, P. 111-116, 1965] Glyceride Structure
[ Agr. Biol. Chem., Vol. 29, No. 2, p. 111-116, 1965] Glyceride Structure and Biosynthesis of Natural Fats Part IV Biosynthetic Process of Triglycerides in Maturing Seed of Chinese Tallow Tree By Osamu HIRAYAMAand Shingo OHAMA Collegeof Agriculture,Kyoto Prefectural University, Kyoto ReceivedAugust 20, 1964 Changes in lipid classes and water-soluble components in maturing seed of Chinese tallow tree were examined. Rapid oil production occurred at three stages of seed maturity. Stillingia oil syntheses in seed kern proceeded more rapidly than stillingia tallow on the kern at early stage. Analytical data for components in immature seed suggest that the pathway of seed oil synthesis is the same as Kennedy's pathway. However, the appear ance of monoglycerides indicates the existence of a side pathway. From specific positional distribution of fatty acids in mono-, di-, and triglyceride from immature seed, synthetic process of glyceride structure was discussed. In previous paper,1) biosynthetic process of flowering, and sample number was given as shown triglycerides in maturing soybean seed has in Table 1. been investigated by measuring changes in Extraction and Fractionation of Lipids and the contents and composition of lipid and Water-soluble Components water-soluble components. A similar investi The seeds were extracted two times with hot gation was attemped on maturing seed of acetone under shaking. The extracts were combined,, Chinese tallow tree (Sapium sebiferum Roxb.) in and evaporated to dryness to obtain crude stillingia the present work. Chinese tallow tree ac- tallow. The fat was further refined by re-extraction with diethyl ether and evaporation. The residual seed cumulates solid fat (stillingia tallow) on the kern was crushed and homogenized with a mixture surface of seed kern, and liquid oil (stillingia of chloroform-methanol (2:1, v/v) using homogenizer. -

Physico-Chemical Characteristics and Fatty Acid Profile of Desert Date Kernel Oil
African Crop Science Journal, Vol. 21, Issue Supplement s3, pp. 723 - 734 ISSN 1021-9730/2013 $4.00 Printed in Uganda. All rights reserved ©2013, African Crop Science Society PHYSICO-CHEMICAL CHARACTERISTICS AND FATTY ACID PROFILE OF DESERT DATE KERNEL OIL C.A. OKIA1,2, J. KWETEGYEKA3, P. OKIROR2, J.M. KIMONDO4, Z. TEKLEHAIMANOT5 and J. OBUA6 1World Agroforestry Centre (ICRAF), P. O. Box 26416, Kampala, Uganda 2College of Agricultural and Environmental Sciences, Makerere University, P. O. Box 7062, Kampala, Uganda 3Department of Chemistry, Kyambogo University, P. O. Box 1, Kyambogo, Uganda 4Kenya Forestry Research Institute, P. O. Box 20412-00200, Nairobi, Kenya 5School of Environment, Natural Resources and Geography, Bangor University, Bangor, Gywnedd, LL57 2UW, UK 6The Inter-University Council for East Africa, P. O. Box 7110, Kampala, Uganda Corresponding author: [email protected] ABSTRACT The desert date (Balanites aegyptiaca (L.) Del.) is an indigenous fruit tree, common in the arid and semi-arid lands of Africa. Its fruits, available in the height of the dry season, contain edible pulp which is an important food for both humans and livestock. Balanites kernel is a source of highly regarded edible and medicinal oil. Both the fruits and oil are trade items in the west Nile sub-region of Uganda. Because of its growing importance as a source of food and income for dryland communities, an assessment of the physico-chemical characteristics and fatty acid profile of kernel oil in Uganda was carried out. Balanites fruit samples were collected from Katakwi, Adjumani and Moroto districts; representing the Teso, West Nile and Karamoja tree populations, respectively. -

64Th Tobacco Science Research Conference
PROGRAM BOOKLET AND ABSTRACTS Volume 64 64th Tobacco Science Research Conference October 3-6, 2010 Hilton Head Island, South Carolina USA Hosts: Cerulean & Global Laboratory Services TOWN OFTO HIWNLT OFON HI HEADLTON ISLAND HEAD ISLAND One Town CenterOne Court, Town Hilton Center Head Court, Island, Hilton S.C. Head 29928 Island, S.C. 29928 (843) 341-4600(843) Fax 341-4600 (843) 842- 7728 Fax (843) 842-7728 www.hiltonheadwwwislandsc.gov.hiltonhe adislandsc.gov Thomas D. Peeples Thomas D. Peeples October 3 – 6, 2010 Mayor Mayor Kenneth S. Heitzke Kenneth S. Heitzke Mayor ProTem Mayor ProTem Council Members Council Members 64th TSRC: “Tobacco Research in the Era of Biotechnology and Willie (Bill) Ferguson Willie (Bill) Ferguson William D. Harkins Genomics” William D. Harkins Drew A. Laughlin John Safay Drew A. Laughlin George W. Williams, JohnJr. Safay Dear TSRC Delegates: George W. Williams, Jr. Stephen G. Riley Town Manager Stephen G. Riley Hilton Head Island is honored to have been chosen as the location for Town Manager your Conference. We welcome the approximately 300 scientists from over a dozen countries who will be in attendance representing all disciplines of the tobacco industry: academia, government agencies and health organizations. Hilton Head Island is widely known as a major convention area, and a world renowned destination location. You will find fine dining, entertainment, golf, tennis, and of course many miles of beautiful beaches to provide you with a delightful visit. The Island is also a unique and diverse community because of its rich, historical heritage and the contributions of the Gullah Culture. The preservation of this culture remains a priority for all of us. -

Influence of the Positions of Unsaturated Acyl Groups in Glycerides on Autoxidation
Agric. Biol Chem., 47 (10), 2251 -2255, 1983 2251 Influence of the Positions of Unsaturated Acyl Groups in Glycerides on Autoxidation Dong Ki Park, Junji Terao and Setsuro Matsushita Research Institute for Food Science, Kyoto University, Uji, Kyoto 611, Japan Received March 3, 1983 The influence of the positions ofunsaturated acyl groups in the glycerides on autoxidation was analyzed in relation to synthesized and soybean oil triglycerides. No discrepancies in rates of autoxidation of unsaturated acyl groups at different positions in the glycerides (between PLP and PPL, between PLnP and PPLn) was observed. Likewise, no discrepancies were observed before or after interesterification of synthesized triglyceride mixtures or soybean oil triglyceride. In the case of trilinolein, peroxidation occurred at random at both the a- and ^-positions. (P, palmitic acid; L, linoleic acid; Ln, linolenic acid.) Little has been reported about the factors in effect of the positions of unsaturated acyl glyceride structures which affect autoxidation. groups in glycerides on autoxidation with the Raghuveer and Hammond1* have reported use of synthesized triglycerides (1,3-dipal- that the stability of most fats decreases after mitoyl-2-olein and 1,2-dipalmitoyl-3-olein; randomization and proposed a theory based 1,3-dipalmitoyl-2-linolein and 1,2-dipalmi- on the hexagonal packing of the acyl chain in toyl-3-linolein; l ,3-dipalmitoyl-2-linolenin and glycerides in the molton state. They suggested l,2-dipalmitoyl-3-linolenin). The effect of in- that acyl groups at the Sn-1 and Sn-3 positions teresterification of triglyceride on autoxidation in glyceride are oxidized more rapidly than was also analyzed. -

Rxi-65TG Columns
Restek GC Accurate, Reliable GC Analysis of Triglycerides in Edible Oils Fight Food Fraud with Rxi-65TG Columns • High-temperature stability ensures consistent results and fewer column changes. • Separate and quantify critical triglycerides without Dependable interference from column bleed. results even after extended periods • Observe even underivatized mono- and diglycerides. at 370 ˚C! www.restek.com Rxi-65TG Columns Ensure Accurate, Reliable GC Analysis of Triglycerides in Edible Oils • High temperature stability ensures consistent results and longer column lifetimes. • Separate and quantify critical triglycerides without interference from column bleed. • Observe even underivatized mono- and diglycerides. Edible oils, especially olive oil, are big business. It’s why honest producers strive for a quality product, and it’s why others cheat consumers by selling adulterated goods that have been blended with or completely replaced by cheaper, lower-quality oils. To protect the industry, food scientists require analytical solutions that dependably determine quality and authenticity. For decades, GC columns with 65% phenyl-substituted polysiloxane stationary phases (65-type) have been used to analyze triglycerides (acylg- lycerols) in edible oils. But, quality and consistency vary significantly because evenly coating these phases inside column tubing is very difficult to do. As a result, 65-type columns can exhibit high bleed and low inertness. Bleed interferes with accurate identification and quantitation, and over time it leads to shifts in retention time, loss of resolution, and poor peak shape due to increasing column activity. The relatively high temperatures used in most triglyceride methods only exacerbate these problems. To provide a better option for food scientists around the world, Restek developed a 65-type column that couples a new high phenyl column-coating technology with the proven manufacturing techniques used to make our premier Rxi column family. -

New Oilseed Crops on the Horizon!
New Oilseed Crops on the Horizon!: L. H. PRINCEN2 Fats and oils for food uses are now plentiful on a worldwide basis. Tallow, lard and fish oils, as well as vegetable oils, such as those derived from soybean, sun 1\ flower, palm, rapeseed, peanut and cottonseed, are often overproduced. Although many ofthese products are also used for industrial chemicals, they often are not [ ofthe most favorable composition for nonfood applications. A search for new oil 1\ seed crops with more advantageous oil composition has led to the development of l- excellent candidates that are now close to commercial acceptance. Among them are Crambe, Limnanthes, Vernonia, Sapium and Simmondsia. Other crops are at a much lower stage ofdevelopment but also have excellent potential. They include Cuphea, Foeniculum, Stokesia, Lesquerella and Lunaria. In this age ofsearching for renewable resources to replace petrochemicals and importedstrategic materials, c a well-organized research and development program on new oilseed crops could u soon result in American self-sufficiency for industrial oils andfatty acids. c Animal fats and vegetable oils make up an important part of the world's ag t ricultural production. Previous papers in this symposium have shown their eco a nomic impact from production through utilization. Although through the ages fats and oils have been produced mainly for food purposes, even centuries ago many also found use in such items as lubricants and greases, paints, soaps, lamp a oil and candles. Today, in the era ofpetrochemicals, fats and oils still playa major ( role in the production ofnonfood materials (Pryde, 1979). For example, in paints t and other coatings, where water-based latex systems have replaced much of the traditional vegetable oil-based paint market, approximately one-third ofthe bind ers used is still based on vegetable oils or their derivatives. -

Biodiesel Production Using Supported 12-Tungstophosphoric Acid As
BIODIESEL PRODUCTION USING SUPPORTED 12-TUNGSTOPHOSPHORIC ACID AS SOLID ACID CATALYSTS A Thesis Submitted to the College of Graduate Studies and Research In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy In the Department of Chemical and Biological Engineering University of Saskatchewan Saskatoon By CHINMOY BAROI Copyright © Chinmoy Baroi, December 2014 All Rights Reserved PERMISSION TO USE In presenting this thesis in partial fulfilment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by Dr. Ajay K. Dalai who supervised my thesis work, or in his absence, by the Dean of the College of Engineering. It is understood that any copying or publication or use of this thesis or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or part should be addressed to: Head of the Department Department of Chemical and Biological Engineering University of Saskatchewan 57 Campus Drive Saskatoon, Saskatchewan Canada S7N 5A9 i ABSTRACT Biodiesel has achieved worldwide recognition for many years due to its renewability, lubricating property, and environmental benefits. -

Characteristics, Composition and Oxidative Stability of Lannea Microcarpa Seed and Seed Oil
Molecules 2014, 19, 2684-2693; doi:10.3390/molecules19022684 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Characteristics, Composition and Oxidative Stability of Lannea microcarpa Seed and Seed Oil Patrice Bazongo 1, Imaël Henri Nestor Bassolé 1,*, Søren Nielsen 2, Adama Hilou 3, Mamoudou Hama Dicko 1 and Vijai K. S. Shukla 2 1 Laboratoire de Biochimie Alimentaire, Enzymologie, Biotechnologie Industrielle et Bioinformatique (Laboratoire BAEBIB), Department of Biochemistry and Microbiology, Université de Ouagadougou, Ouagadougou 03 03 BP 7021, Burkina Faso; E-Mails: [email protected] (P.B.); [email protected] (M.H.D.) 2 International Food Science Centre (IFSC A/S), Sønderskovvej, Lystrup 7 DK-8520, Denmark; E-Mails: [email protected] (S.N.); [email protected] (V.K.S.S.) 3 Laboratoire de Biochimie et Chimie Appliquées (LABIOCA), UFR/SVT, Université de Ouagadougou, Ouagadougou 09 09 BP 848, Burkina Faso; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +226-7812-5004. Received: 13 November 2013; in revised form: 6 December 2013 / Accepted: 9 December 2013 / Published: 24 February 2014 Abstract: The proximate composition of seeds and main physicochemical properties and thermal stability of oil extracted from Lannea microcarpa seeds were evaluated. The percentage composition of the seeds was: ash (3.11%), crude oil (64.90%), protein (21.14%), total carbohydrate (10.85%) and moisture (3.24%). Physicochemical properties of the oil were: refractive index, 1.473; melting point, 22.60°C; saponification value, 194.23 mg of KOH/g of oil; iodine value, 61.33 g of I2/100 g of oil; acid value, 1.21 mg of KOH/g of oil; peroxide value, 1.48 meq of O2/kg of oil and oxidative stability index, 43.20 h. -

A-Anions 221 Acetate 23, 106 Acetate-Malonate Pathway 24
Index a-anions 221 anti-block agent 218, 237 acetate 23, 106 antioxidants 84, 173, 175, 176 acetate-malonate pathway 24 anti-slip agent 218, 237 acetylenic acids 10, 11, 159 Apis mellifera 85 acetylenic intermediates 30 apricot oil 66, 67 acid anhydrides 214 arachidic acid 5, 6 acid chlorides 214 arachidonic acid 8,9, 29, 184 acidity 102 synthesis 31, 32 acidolysis 208,209, 213 Arachis hypogaea 67 ACP, see acyl carrier protein argentation tic 19 activated carbon 90 arsenites 18 acyl carrier protein 25 asci epic acid 7 acylation 47 Aspergillus niger 88, 97, 213 acylglycerols 36, 52 Aspergillus oryzae 213 acylpyrrolidide 154 auricolic acid 13, 14 alchornoic acid 15 autoxidation 162-167 alcohols 217,238 avenasterol 84 alcoholysis 208, 209 avocado oil 67 alepramic acid 14 aziridines 202 aleprestic acid 14 alepric acid 14 babassu oil 88 aleprolic acid 14 beef tallow 72 aleprylic acid 14 beeswax 84 Aleurites fordii 10 behenic acid 5, 6 alkanoic acids, crystal structure 130 bentonitite 90 alkylbenzoxazole 154 benzyl-sn-glycerol 54 alkyl dimethyloxazole 154 benzylidene glycerol 48 alkyl ketene dimers (AKD) 215 Betapol 55, 214 alkyl polyglucoside 217 BHA, see butylated hydroxyanisole allenic acids 10, 11, 39 BHT, see butylated hydroxy toluene allenic epoxides 187 biodiesel 241 allylic bromination 194 biohydrogenation 161 alklylic hydroperoxides 162, 163, 165 biosynthesis 24, 44 almond oil 66, 67, 69 blackcurrant oil 64, 65 alternation 130 bleaching 89, 90 amides 218, 238 Bligh and Dyer method 101 amine oxides 219, 220, 237 BOB, see oleodibehenin -
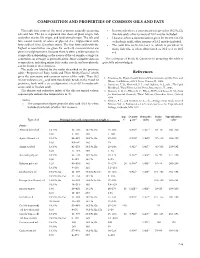
Composition and Properties of Common Oils and Fats References
COMPOSITION AND PROPERTIES OF COMMON OILS AND FaTS This table lists some of the most common naturally occurring • In some oils where a concentration is given for 18:2 9c,12c oils and fats . The list is separated into those of plant origin, fish (linoleic acid), other isomers of 18:2 may be included . and other marine life origin, and land animal origin . The oils and • Likewise, where a concentration is given for 18:3 9c,12c,15c fats consist mainly of esters of glycerol (i .e ., triglycerides) with (α-linolenic acid), other isomers of 18:3 may be included . fatty acids of 10 to 22 carbon atoms . The four fatty acids with the • The acid 20:5 6c,9c,12c,15c,17c, which is prevalent in highest concentration are given for each oil; concentrations are many fish oils, is often abbreviated as 20:5 ω-3 or 20:5 given in weight percent . Because there is often a wide variation in n-3 . composition depending on the source of the oil sample, a range (or sometimes an average) is generally given . More complete data on The assistance of Frank D . Gunstone in preparing this table is composition, including minor fatty acids, sterols, and tocopherols, gratefully acknowledged . can be found in the references . The acids are labeled by the codes described in the previous table, “Properties of Fatty Acids and Their Methyl Esters,” which References gives the systematic and common names of the acids . Thus 18:2 1 . Firestone, D ., Physical and Chemical Characteristics of Oils, Fats, and 9c,12c indicates a C18 acid with two double bonds in the 9 and 12 Waxes, 2nd Edition, AOCS Press, Urbana, IL, 2006 .