The Asymptomatic Outpatient with Abnormal Liver Function Tests 169 Leading to Shortage of Pyridoxal 50-Phosphate, Which Is a Cofactor for Both AST and ALT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

Liver Function Tests in Type 2 Sudanese Diabetic Patients
International Journal of Nutrition and Metabolism Vol. 3(2) pp. 17-21, February 2011 Available online http://www.academicjournals.org/ijnam ISSN 2141-2340 ©2011 Academic Journals. Full Length Research Paper Liver function tests in type 2 Sudanese diabetic patients Ayman S. Idris 1*, Koua Faisal Hammad Mekky 1, Badr Eldin Elsonni Abdalla 2 and Khalid Altom Ali 3 1Department of Biochemistry, Faculty of Science and Technology, Al-Neelain University, P. O. Box 12702, Al Baladya St. Khartoum, Sudan. 2Department of Biochemistry, Faculty of Medicine, University of Gezira, Sudan. 3Department of Biochemistry, Faculty of Medicine, International University of Africa, Sudan. Accepted 21 January, 2011 This study was planned to evaluate the liver function in patients with type 2 diabetes mellitus by measuring aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma glutamyl transpeptidase (GGT), total protein and albumin. The study was carried out in Wad Medani, Abo Agla Diabetes Centre. 50 patients with type 2 diabetes mellitus (23 male and 27 female) were included in the study. Their ages ranged between 43 and 79 years. Thirty matched normal individuals were taken as control group. In the present study, mean values of ALT, AST and GGT were significantly higher in patients than in the control (P<0.001). Total protein and albumin concentrations in patients were lower compared to control group (P<0.01). The mean of serum glucose in patients revealed significant difference (P<0.000) in comparison to the control group. Although the differences were statistically significant, the means of ALT, AST, GGT, total protein and albumin were falling within the normal range. -
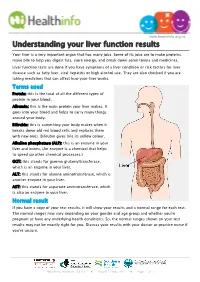
Understanding Your Liver Function Results
www.healthinfo.org.nz Understanding your liver function results Your liver is a very important organ that has many jobs. Some of its jobs are to make proteins, make bile to help you digest fats, store energy, and break down some toxins and medicines. Liver function tests are done if you have symptoms of a liver condition or risk factors for liver disease such as fatty liver, viral hepatitis or high alcohol use. They are also checked if you are taking medicines that can affect how your liver works. Terms used Protein: this is the total of all the different types of protein in your blood. Albumin: this is the main protein your liver makes. It goes into your blood and helps to carry many things around your body. Bilirubin: this is something your body makes when it breaks down old red blood cells and replaces them with new ones. Bilirubin gives bile its yellow colour. Alkaline phosphatase (ALP): this is an enzyme in your liver and bones. (An enzyme is a chemical that helps to speed up other chemical processes.) GGT: this stands for gamma glutamyltransferase, which is an enzyme in your liver. ALT: this stands for alanine aminotransferase, which is another enzyme in your liver. AST: this stands for aspartate aminotransferase, which is also an enzyme in your liver. Normal result If you have a copy of your test results, it will show your results and a normal range for each test. The normal ranges may vary depending on your gender and age group and whether you're pregnant or have any underlying health conditions. -

Laboratory Tests Haematology
LABORATORY TESTS HAEMATOLOGY TEST FUNCTION UNITS INCREASED DECREASED Hb Oxygen carrying component of blood g/dL Dehydration Blood loss Haemoglobin Chronic obstructive lung disease After anticancer drugs eg chemotherapy & Smoking PARP inhibitors, due to bone marrow Heart failure depression Renal cancer Iron, folate and vitamin B12 deficiency Haematological malignancy Chronic illness Haemolysis Chronic kidney disease Haematological malignancy Platelets (Thrombocytes) Vital for blood coagulation. 109/L Thrombocythaemia Thrombocytopenia Acute blood loss Infections Chronic illness Drugs – e.g. cytostatics Certain forms of anaemia Radiotherapy (rare) Infection Immunologic disorders Poor spleen function Haematological malignancy Cancer infiltrating bone marrow 9 WBC Protect the body against invading micro- 10 /L Infections Drug – e.g. cytostatics White Blood Cells / Leukocytes organisms. Haematological malignancies Radiotherapy (rare) Other cancers Haematological malignancy Differential: Basophils The relative percentage of the various Therapy with corticosteroids Cancer infiltrating bone marrow Eosinophils cells found in the blood is known as the Metabolic illnesses Immune disorders Neutrophils (white cell) differential count. Recovering bone marrow Severe infections Lymphocytes Monocytes ESR Non-specific indication of inflammation mm/H Focus or cause of inflammation Immune disorders Erythrocyte Sedimentation Infection Congestive heart failure Rate Connective tissue disorder Rheumatoid arthritis -

Liver Function Testing
Liver function testing What are liver function tests? Role of the liver A liver function test (LFT) is a blood test that gives an What are liver function tests? indication of whether the liver is functioning properly. The test is also very useful to see if there is active What are the substances damage in the liver (hepatitis) or sluggish bile flow (cholestasis). measured in an LFT blood test and what’s so important about Liver function tests measure the amount of particular them? chemicals in the blood. This gives a gauge of possible damage to liver cells - damage that can be caused by Total protein many things including HCV. So a more correct term for Albumin a liver test would actually be a liver dysfunction test. Globulin It’s important to remember that diagnosis of liver Bilirubin disease depends on a combination of patient history, physical examination, laboratory testing, biopsy and GGT sometimes imaging studies such as ultrasound scans. ALK Phos Diagnosis of hepatitis C usually also involves antibody tests (see Edition 19) or PCR tests (see Edition 20). ALT AST People reading this page should keep in mind that abnormalities within liver tests don’t necessarily point Platelet count to specific diseases. Only a physician who knows all Adult range / normal range the aspects of a specific case can reliably make a diagnosis. What are the substances measured in an LFT blood test and what’s so important about them? Role of the liver Total protein The role of the liver is to keep the body’s complex internal chemistry in balance. -

Evaluation of Thyroid Function Tests in Children with Chronic Liver Diseases
ORIGINAL ARTICLE DOI: 10.4274/jcrpe.galenos.2019.2019.0029 J Clin Res Pediatr Endocrinol 2020;12(2):143-149 Evaluation of Thyroid Function Tests in Children with Chronic Liver Diseases Ş. Şebnem Ön1, Sezer Acar2, Korcan Demir2, Ayhan Abacı2, Yeşim Öztürk3, Sinem Kahveci Çelik3, Ece Böber2 1Dokuz Eylül University Faculty of Medicine, Department of Pediatrics, İzmir, Turkey 2Dokuz Eylül University Faculty of Medicine, Department of Pediatric Endocrinology, İzmir, Turkey 3Dokuz Eylül University Faculty of Medicine, Department of Pediatric Gastroenterology, İzmir, Turkey What is already known on this topic? Thyroid hormone metabolism may be impaired in adult chronic liver diseases and subclinical hypothyroidism (SH) (≈3.5%) or euthyroid sick syndrome (7-30%) may occur. There are very few studies in the pediatric age group investigating this issue. What this study adds? The proportion of pediatric/adolescent chronic liver disease patients with SH or euthyroid sick syndrome was around 10%. It should be kept in mind that children with chronic liver disease may have SH. Abstract Objective: Studies examining changes in thyroid function in the course of chronic liver disease have mostly been conducted in adults. The aim of this study was to investigate thyroid dysfunction in children with chronic liver diseases. Methods: Between 2005 and 2018, patients aged up to 18 years of age, diagnosed with chronic liver disease and had thyroid function test results available were included. Anthropometric characteristics, liver and thyroid function results were collected and analyzed. Results: The study included 107 (53 female; 49.5%) patients aged between one month and 18 years-old. -

Interpreting Liver Function Tests CONTENTS
A CASE-BASED SERIES ON PRACTICAL PATHOLOGY FOR GPs FEBRUARY 2020 Interpreting liver function tests CONTENTS: • Dealing with abnormal LFTs in the asymptomatic patient • Diagnosing Gilbert’s syndrome © The Royal College of • Establishing the source of raised ALP Pathologists of Australasia 2 Authors: Dr Melissa Gillett Dr Rebecca Brereton MBBS, FRACP, FRCPA, MAACB Specialist Chemical Pathologist, Chemical Pathologist, Fiona Stanley Hospital Network Fiona Stanley Hospital Network Laboratory, PathWest Laboratory Laboratory, PathWest Laboratory Medicine, Murdoch, WA Medicine, Murdoch, WA Common Sense Pathology is developed by the Royal College of Pathologists of Australasia and supported by Australian Doctor Group. © 2020 Royal College of Pathologists of Australasia www.rcpa.edu.au CEO: Dr Debra Graves Email: [email protected] While the views expressed are those of the authors, modified by expert reviewers, they are not necessarily held by the College. Published by Australian Doctor Group Level 2, 26-32 Pyrmont Bridge Road, Pyrmont NSW 2009 Ph: 1300 360 126 Email: [email protected] Website: www.australiandoctorgroup.com.au ACN: 615 959 914 ABN: 94 615 959 914 ISSN: 1039-7116 The views expressed in this publication are not necessarily those of Australian Doctor Group. This issue is produced and owned by the Royal College of Pathologists of Australasia and distributed by Australian Doctor Group. Common Sense Pathology Editor: Dr Steve Flecknoe-Brown Email: [email protected] Editor: Dr Karley Heyworth Email: [email protected] Sub-editor: Lesley Hoye Email: [email protected] Graphic Designer: Kate O’Dea Email: [email protected] For an electronic version of this issue, please visit www.howtotreat.com.au You can also visit the Royal College of Pathologists of Australasia’s website at www.rcpa.edu.au Click on Library and Publications, then Common Sense Pathology. -
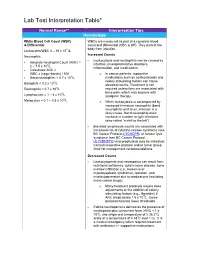
Lab Test Interpretation Table*
Lab Test Interpretation Table* Normal Range** Interpretation Tips Hematology White Blood Cell Count (WBC) WBCs are measured as part of a complete blood & Differential count and differential (CBC & diff). They protect the body from infection. Leukocytes/WBC 4 – 10 x 109 /L Increased Counts Neutrophils - Leukocytosis and neutrophilia can be caused by Absolute Neutrophil Count (ANC) = - infection, myeloproliferative disorders, 2 – 7.5 x 109/L inflammation, and medications. - Calculated ANC = WBC x (segs+bands) / 100 o In cancer patients, supportive - Band neutrophils: < 0.7 x 109/L medications such as corticosteroids and 9 colony stimulating factors can cause Basophils < 0.2 x 10 /L elevated counts. Treatment is not Eosinophils < 0.7 x 109/L required unless they are associated with 9 bone pain, which may improve with Lymphocytes = 1 – 4 x 10 /L analgesic therapy. 9 Monocytes = 0.1 – 0.8 x 10 /L o When leukocytosis is accompanied by increased immature neutrophils (band neutrophils) and fever, infection is a likely cause. Band neutrophils often increase in number to fight infections (also called “a shift to the left”). - Elevated lymphocyte counts are associated with increased risk of cytokine-release syndrome (see BC Cancer Protocol LYCHOPR) or tumour lysis syndrome (see BC Cancer Protocol ULYVENETO) and prophylaxis may be indicated. Consult respective protocol and/or tumor group chair for management recommendations. Decreased Counts - Leukocytopenia and neutropenia can result from nutritional deficiency, autoimmune disease, bone marrow infiltration (i.e., leukemia or myelodysplastic syndrome), radiation, and myelosuppression due to medications (including many cancer drugs). o Many treatment protocols require dose adjustments or the addition of colony stimulating factors (e.g., filgrastim) if ANC drops below 1.5 x 109/L. -

ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries
18 PRACTICE GUIDELINES CME ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries P a u l Y. K w o , M D , F A C G , F A A S L D 1 , Stanley M. Cohen , MD, FACG, FAASLD2 and Joseph K. Lim , MD, FACG, FAASLD3 Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defi ned as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defi ned as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. The evaluation of hepatocellular injury includes testing for viral hepatitis A, B, and C, assessment for nonalcoholic fatty liver disease and alcoholic liver disease, screening for hereditary hemochromatosis, autoimmune hepatitis, Wilson’s disease, and alpha-1 antitrypsin defi ciency. In addition, a history of prescribed and over-the-counter medicines should be sought. For the evaluation of an alkaline phosphatase elevation determined to be of hepatic origin, testing for primary biliary cholangitis and primary sclerosing cholangitis should be undertaken. -

Interpreting Liver Function Tests
Med/Psych Update pSYCHIATRY Tonya L. Fancher, MD, MPH Interpreting liver function tests Assistant professor Department of internal medicine Amit Kamboj, MD Medicine resident Department of internal medicine Patients’ elevated LFT results can indicate John Onate, MD Assistant professor hepatocyte injury, cholestasis, or both Department of psychiatry and human behavior rs. W, age 53, is referred by her Interpreting psychiatric patients’ liver University of California, Davis Medical primary provider for consultation function tests (LFTs) can be challenging, Center on depressive symptoms, including Sacramento M ® Dowdenespecially in Healththose with polypharmacy,Media worsening depressed mood, anhedonia, co-occurring substance abuse, or risk fac- anxiety, and suicidal thoughts for 2 months. tors for viral hepatitis. You can improve She reports at least 2 similarCopyright episodes Forin the personalcollaboration withuse primary only care providers past 15 years. Mrs. W has a remote history by understanding: of IV drug use and history of alcohol abuse, • what an LFT measures but she attends Alcoholics Anonymous and • how to interpret abnormal results has 10 years of sobriety. She has no history • which conditions to suspect, based on of hospitalizations for medical illness and the results. denies any medical problems. A standard LFT usually measures sev- Mrs. W is taking amitriptyline, 50 mg, eral enzymes and proteins, typically ALT, for insomnia. She has no history of manic AST, alkaline phosphatase (ALP), total or psychotic symptoms, and the mental bilirubin (TBIL), albumin (ALB), and total status examination is consistent with major protein (TP). Measures of gamma-glutamyl depression. Her past depressive episodes transpeptidase (GGT) and prothrombin were treated successfully with a medication time (PT) are often requested with an LFT. -

Nutritional Evaluation of Serum Lactate Dehydrogenase Levels in Humans
〔541〕 Nutritional Evaluation of Serum Lactate Dehydrogenase Levels in Humans Masahide Imaki, Tamotsu Miyoshi and Takeshi Yoshimura Department of Public Health, School of Medicine, University of Tokushima, Tokushima INTRODUCTION Measurements of the activity and isozyme pattern of serum lactate dehydrogenase (LDH) have been widely used as liver function tests. The serum LDH activity is often increased in heart or liver diseases1,2), and the changes in the serum LDH isozyme pattern reflect characteristic disturbances in various organs. Serum gamma-glutamyl transpeptidase is used as an index of alcohol consumption3) in healthy people, but serum LDH activity and the LDH isozyme pattern are not used as health indices. Since the serum LDH activity and LDH isozyme pattern would be easy to measure in screening tests, we examined the availability of use of these parameters as health indices by studies on their changes with nutrient intake. METHODS This study consisted of two parts, an epidemiological study and a laboratory study. Epidemiological study: A total of 102 volunteers were examined. The volunteers included 92 men Table 1 Characteristics of the subjects in the and 10 women between the ages of 20 and 28 who had epidemiological study. no past history of liver trouble. Table 1 shows data on the physical characteristics of the subjects. Blood was usually collected from the cubital vein of subjects after over-night fasting. The serum was separated, and the serum LDH activity and per- centages of LDH isozymes were measured on the same day. Nnmber of subjects: 102 (92men, 10women), Rohrer Serum LDH activity was determined by a UV index: (Weight/Right3)×107. -

Evaluation of Abnormal Liver Function Tests J K Limdi, G M Hyde
307 REVIEW Postgrad Med J: first published as 10.1136/pmj.79.932.307 on 1 June 2003. Downloaded from Evaluation of abnormal liver function tests J K Limdi, G M Hyde ............................................................................................................................. Postgrad Med J 2003;79:307–312 Interpretation of abnormalities in liver function tests is a sterile ear or body piercing, blood or blood product common problem faced by clinicians. This has become transfusions), medications used currently or pre- viously, herbal or alternative remedies, and occu- more common with the introduction of automated pational exposure to toxins (box 2). routine laboratory testing. Not all persons with one or Other factors such as diabetes, obesity and more abnormalities in these tests actually have liver hyperlipidaemia in non-alcoholic fatty liver dis- ease, and family history (for Wilson’s disease, disease. The various biochemical tests, their haemochromatosis, autoimmune disease) may be pathophysiology, and an approach to the interpretation significant. of abnormal liver function tests are discussed in this review. BILIRUBIN .......................................................................... Bilirubin is formed from the lysis of red cells (the haem component) within the reticuloendothelial system. Unconjugated bilirubin is transported to ommonly available tests include alanine the liver loosely bound to albumin. It is water transaminase (ALT) and aspartate insoluble and therefore cannot be excreted in Ctransaminase (AST), alkaline phosphatase urine. Conjugated bilirubin is water soluble and (ALP), gammaglutamyl transferase, serum bi- appears in urine. lirubin, prothrombin time, or international nor- Within the liver it is conjugated to bilirubin malised ratio and serum albumin (box 1). They glucoronide and subsequently secreted into bile reflect different functions of the liver—that is, to and the gut respectively.