The Effect of Functional Fiber on Microbiota Composition in Different Intestinal Segments of Obese Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ecology and Epidemiology of Campylobacter Jejuni in Broiler Chickens
Ecology and Epidemiology of Campylobacter jejuni in Broiler Chickens A DISSERTATION SUBMITTED TO THE FACULTY OF THE UNIVERSITY OF MINNESOTA BY Hae Jin Hwang IN PARTIALL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Dr. Randall Singer, Dr. George Maldonado June 2019 © Hae Jin Hwang, 2019 Acknowledgements I would like to sincerely thank my advisor, Dr. Randall Singer, for his intellectual guidance and support, great patience, and mentorship, which made this dissertation possible. I would also like to thank Dr. George Maldonado for his continuous encouragement and support. I would further like to thank my thesis committee, Dr. Richard Isaacson and Dr. Timothy Church, for their guidance throughout my doctoral training. I thank all my friends and colleagues I met over the course of my studies. I am especially indebted to my friends, Dr. Kristy Lee, Dr. Irene Bueno Padilla, Dr. Elise Lamont, Madhumathi Thiruvengadam, Dr. Kaushi Kanankege and Dr. Sylvia Wanzala, for their support and friendship. Heartfelt gratitude goes to my family, for always believing in me, encouraging me and helping me get through the difficult and stressful times during my studies. Lastly, I thank Sven and Bami for being the best writing companions I could ever ask for. i Abstract Campylobacteriosis, predominantly caused by Campylobacter jejuni, is a common, yet serious foodborne illness. With consumption and handling of poultry products as the most important risk factor of campylobacteriosis, reducing Campylobacter contamination in poultry products is considered the best public health intervention to reduce the burden and costs associated with campylobacteriosis. To this end, there is a need to improve our understanding of epidemiology and ecology of Campylobacter jejuni in poultry. -

Comparison of Intestinal Permeability, Morphology, and Ileal Microbial Communities of Commercial Hens Housed in Conventional Cages and Cage-Free Housing Systems
Animal Science Publications Animal Science 2020 Comparison of intestinal permeability, morphology, and ileal microbial communities of commercial hens housed in conventional cages and cage-free housing systems Maddison L. Wiersema Iowa State University, [email protected] Lucas Koester Iowa State University, [email protected] Stephan Schmitz-Esser Iowa State University, [email protected] Dawn A. Koltes Iowa State University, [email protected] Follow this and additional works at: https://lib.dr.iastate.edu/ans_pubs Part of the Microbial Physiology Commons, Poultry or Avian Science Commons, and the Veterinary Microbiology and Immunobiology Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ ans_pubs/618. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Animal Science at Iowa State University Digital Repository. It has been accepted for inclusion in Animal Science Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Comparison of intestinal permeability, morphology, and ileal microbial communities of commercial hens housed in conventional cages and cage-free housing systems Abstract The gastrointestinal health of poultry can be impacted by a variety of factors including their environment. As egg production moves from conventional cage housing (CC) towards cage-free housing (CF), it is important to understand this impact on intestinal health. This study was conducted to determine if housing type impacted intestinal permeability, morphology, and microbial communities in commercial hens across housing systems. -

Reorganising the Order Bacillales Through Phylogenomics
Systematic and Applied Microbiology 42 (2019) 178–189 Contents lists available at ScienceDirect Systematic and Applied Microbiology jou rnal homepage: http://www.elsevier.com/locate/syapm Reorganising the order Bacillales through phylogenomics a,∗ b c Pieter De Maayer , Habibu Aliyu , Don A. Cowan a School of Molecular & Cell Biology, Faculty of Science, University of the Witwatersrand, South Africa b Technical Biology, Institute of Process Engineering in Life Sciences, Karlsruhe Institute of Technology, Germany c Centre for Microbial Ecology and Genomics, University of Pretoria, South Africa a r t i c l e i n f o a b s t r a c t Article history: Bacterial classification at higher taxonomic ranks such as the order and family levels is currently reliant Received 7 August 2018 on phylogenetic analysis of 16S rRNA and the presence of shared phenotypic characteristics. However, Received in revised form these may not be reflective of the true genotypic and phenotypic relationships of taxa. This is evident in 21 September 2018 the order Bacillales, members of which are defined as aerobic, spore-forming and rod-shaped bacteria. Accepted 18 October 2018 However, some taxa are anaerobic, asporogenic and coccoid. 16S rRNA gene phylogeny is also unable to elucidate the taxonomic positions of several families incertae sedis within this order. Whole genome- Keywords: based phylogenetic approaches may provide a more accurate means to resolve higher taxonomic levels. A Bacillales Lactobacillales suite of phylogenomic approaches were applied to re-evaluate the taxonomy of 80 representative taxa of Bacillaceae eight families (and six family incertae sedis taxa) within the order Bacillales. -

Name: Yaniella Halotolerans Authors: (Li Et Al. 2004) Li Et Al. 2008
Compendium of Actinobacteria from Dr. Joachim M. Wink University of Braunschweig Name: Yaniella halotolerans Authors: (Li et al. 2004) Li et al. 2008 Status: comb. nov. Reference: Int. J. Syst. Evol. Microbiol. 58:526 Risk group: 1 (German classification) Type strain: CCTCC AA 001023, DSM 15476, JCM 13527, YIM 70085 Synonym(s): Yania halotolerans (basonym) Author(s) Li WJ;Chen HH;Xu P;Zhang YQ;Schumann P;Tang SK;Xu LH;Jiang CL Title Yania halotolerans gen. nov., sp. nov., a novel member of the suborder Micrococcineae from saline soil in China Journal Int J Syst Evol Microbiol Volume 54 Page(s) 525-531 Year 2004 Microplate with ISP- and melanin media Copyright: PD Dr. Joachim M. Wink, HZI - Helmholtz-Zentrum für Infektionsforschung GmbH, Inhoffenstr. 7, 38124 Braunschweig, Germany, Mail: [email protected]. Compendium of Actinobacteria from Dr. Joachim M. Wink University of Braunschweig Genus: Yaniella FH 6874 Species: halotolerans Numbers in other collections: DSM 15476 Morphology: G R ISP 2 good signal yellow A SP none none G R ISP 3 none A SP G R ISP 4 none A SP G R ISP 5 none A SP G R ISP 6 good signal yellow A SP none none G R ISP 7 none A SP Melanoid pigment: - - - - NaCl resistance: % Lysozyme resistance: pH: Value- Optimum- Temperature : Value- Optimum- 28 °C Carbon utilization: Glu Ara Suc Xyl Ino Man Fru Rha Raf Cel n.d. Enzymes: 2(+) 3+ 4+ 5- 6+ 7(+) 8- 9- 10- 11+ 12+ 13- 14- 15- 16- 17- 18- 19- 20- Nit Pyz Pyr Pal βGur βGal αGlu βNag Esc Ure Gel - - - (+) - - - - - + - Glu Rib Xyl Man Mal Lac Sac Glyg Comments: Good growth on ISP 5 medium with 0,5 % yeast extract and 10 % KCl Copyright: PD Dr. -

An Assessment of Broiler Litter Bacterial Diversity Using Next Generation DNA Sequencing
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 12-2014 An Assessment of Broiler Litter Bacterial Diversity using Next Generation DNA Sequencing Jack Liu Davitt University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Recommended Citation Davitt, Jack Liu, "An Assessment of Broiler Litter Bacterial Diversity using Next Generation DNA Sequencing. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/3146 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Jack Liu Davitt entitled "An Assessment of Broiler Litter Bacterial Diversity using Next Generation DNA Sequencing." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Environmental and Soil Sciences. Shawn Hawkins, Major Professor We have read this thesis and recommend its acceptance: Arnold Saxton, Mark Radosevich, Alice Layton, Mike Smith, Forbes Walker Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) An Assessment of Broiler Litter Bacterial Diversity using Next Generation DNA Sequencing A Thesis Presented for the Master of Science Degree The University of Tennessee, Knoxville Jack Liu Davitt December 2014 Copyright © 2014 by Jack Liu Davitt All rights reserved. -

Changes in the Gut Microbiota During Asian Particolored Bat (Vespertilio Sinensis) Development
Changes in the gut microbiota during Asian particolored bat (Vespertilio sinensis) development Zhongwei Yin1,2, Keping Sun2, Aoqiang Li2, Deyi Sun2, Zhongle Li2,3, Guohong Xiao2 and Jiang Feng1,2 1 College of Life Science, Jilin Agricultural University, Changchun, China 2 Jilin Provincial Key Laboratory of Animal Resource Conservation and Utilization, Northeast Normal University, Changchun, China 3 College of Animal Science and Technology, Jilin Agricultural University, Changchun, China ABSTRACT Background: The gut microbiota is closely linked to host development, diet and health and is influenced by both the host and the environment. Although many studies have focused on the dynamics of the gut microbiota during development in captive animals, few studies have focused on the dynamics of the gut microbiota during development in wild animals, especially for the order Chiroptera. Methods: In this study, we characterized the gut microbiota of the wild Asian particolored bat (Vespertilio sinensis) from 1 day to 6 weeks after birth. We explored the changes in their gut microbial community compositions, examined possible influencing factors, and predicted the feeding transition period. Results: The gut microbiota changed during the development of V. sinensis. The alpha diversity of the bats’ gut microbiota gradually increased but did not change significantly from the 1st day to the 4th week after birth; however, the alpha diversity decreased significantly in week 5, then stabilized. The beta diversity differed slightly in weeks 4–6. In week 4, the fecal samples showed the highest diversity in bacterial community composition. Thus, we predicted that the potential feeding transition period for V. sinensis may occur during week 4. -

Production of Bioagent for Calcium-Based Biocement
60 International Journal of Biotechnology for Wellness Industries, 2016, 5, 60-69 Production of Bioagent for Calcium-Based Biocement Viktor Stabnikov1,2,* 1School of Civil and Environmental Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore 2Department of Microbiology and Biotechnology, National University of Food Technologies, 68 Volodymyrska Street, Kiev 01601, Ukraine Abstract Biocements and biogrouts are developing extensively as new materials alternative to cement and toxic chemical grouts. The most popular type of biocement is a mixture of urease-producing bacteria, urea and calcium salt. Thus, development of biotechnology to produce biomass of urease-active bacteria for large-scale biocementation is an important biotechnological task. Two strains of urease-producing bacteria, Yaniella sp. VS8 and Bacillus sp. VS1 that synthesized inducible and constitutive urease, respectively, were used in the present study. It was shown that low cost biomass of urease-active bacteria can be produced from the hydrolyzed excessive activated sludge of municipal wastewater treatment plant. The biomass of Yaniella sp. VS8 grown in this medium diminished the hydraulic conductivity -4 -8 of sand from 4.8×10 m/s to 5·10 m/s after several biotreatments with solution of 1.5 M urea and 0.75M СаCl2. Keywords Bacillus sp., Yaniella sp., Activated sludge, Biocement, Biogrout. INTRODUCTION The medium ATCC 1376 NH4-YE, which contains yeast extract, 20 g; (NH4)2SO4, 10 g; 0.13 M Tris buffer New area of Biotechnology, Construction (pH 9.0), 1L, is recommended for cultivation of urease- Biotechnology, is developing in two directions: 1) producing bacteria Sporosarcina pasteurii (former biotechnological production of construction materials, Bacillus pasteurii) and is most often used for laboratory and 2) biotechnology of construction processes [1]. -

Genome-Based Taxonomic Classification of the Phylum
ORIGINAL RESEARCH published: 22 August 2018 doi: 10.3389/fmicb.2018.02007 Genome-Based Taxonomic Classification of the Phylum Actinobacteria Imen Nouioui 1†, Lorena Carro 1†, Marina García-López 2†, Jan P. Meier-Kolthoff 2, Tanja Woyke 3, Nikos C. Kyrpides 3, Rüdiger Pukall 2, Hans-Peter Klenk 1, Michael Goodfellow 1 and Markus Göker 2* 1 School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom, 2 Department Edited by: of Microorganisms, Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures, Braunschweig, Martin G. Klotz, Germany, 3 Department of Energy, Joint Genome Institute, Walnut Creek, CA, United States Washington State University Tri-Cities, United States The application of phylogenetic taxonomic procedures led to improvements in the Reviewed by: Nicola Segata, classification of bacteria assigned to the phylum Actinobacteria but even so there remains University of Trento, Italy a need to further clarify relationships within a taxon that encompasses organisms of Antonio Ventosa, agricultural, biotechnological, clinical, and ecological importance. Classification of the Universidad de Sevilla, Spain David Moreira, morphologically diverse bacteria belonging to this large phylum based on a limited Centre National de la Recherche number of features has proved to be difficult, not least when taxonomic decisions Scientifique (CNRS), France rested heavily on interpretation of poorly resolved 16S rRNA gene trees. Here, draft *Correspondence: Markus Göker genome sequences -

Actinobacteriological Research in India
Indian Journal of Experimental Biology Vol. 51, August 2013, pp. 573-596 Review Article Actinobacteriological research in India Sonashia Velho-Pereira & Nandkumar M Kamat* Department of Botany, Goa University, Taleigao Plateau, Goa, 403 206, India Actinobacteria are important sources of compounds for drug discovery and have attracted considerable pharmaceutical, chemical, agricultural and industrial interests. Actinobacteriological research is still in its infancy in India. Early work on actinobacteria started in the 20th century and mostly focused on studying the diversity, identification and screening for antibiotics, enzymes and enzyme inhibitors. Exploration of diverse habitats for the isolation of actinobacteria, have yielded till date 23 novel species. Screening of actinobacteria for antagonistic activity, has led to the discovery of four novel antibiotics. Research on enzymes mostly covered lipases, amylases, proteases, endoglucanases, α-galactosidases, pectin lyases, xylanases, L-asparaginases, L-glutaminase and cellulases. Research on exploiting actinobacteria for other purposes such as production of enzyme inhibitors, single cell protein, bioemulsifier and biosurfactants is still in the experimental stage. This review compiles the work done in last few years, with an emphasis on actinobacterial diversity and bioprospecting for pharmaceutically important compounds like antibiotics, enzymes and other important applications. The chemical creativity and biotechnological potential of Indian actinobacterial strains are yet to be fully -

High Diversity and Suggested Endemicity of Culturable Actinobacteria in an Extremely Oligotrophic Desert Oasis
High diversity and suggested endemicity of culturable Actinobacteria in an extremely oligotrophic desert oasis Hector Fernando Arocha-Garza1, Ricardo Canales-Del Castillo2, Luis E. Eguiarte3, Valeria Souza3 and Susana De la Torre-Zavala1 1 Facultad de Ciencias Biológicas, Instituto de Biotecnología, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Nuevo León, Mexico 2 Facultad de Ciencias Biológicas, Laboratorio de Biología de la Conservación, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Nuevo León, Mexico 3 Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México, Mexico City, Mexico ABSTRACT The phylum Actinobacteria constitutes one of the largest and anciently divergent phyla within the Bacteria domain. Actinobacterial diversity has been thoroughly researched in various environments due to its unique biotechnological potential. Such studies have focused mostly on soil communities, but more recently marine and extreme environments have also been explored, finding rare taxa and demonstrating dispersal limitation and biogeographic patterns for Streptomyces. To test the distribution of Actinobacteria populations on a small scale, we chose the extremely oligotrophic and biodiverse Cuatro Cienegas Basin (CCB), an endangered oasis in the Chihuahuan desert to assess the diversity and uniqueness of Actinobacteria in the Churince System with a culture-dependent approach over a period of three years, using nine selective media. The 16S rDNA of putative Actinobacteria were sequenced using both bacteria universal and phylum-specific primer pairs. Phylogenetic reconstructions were performed to analyze OTUs clustering and taxonomic identification of the isolates in an evolutionary context, using validated type species of Streptomyces from previously phylogenies as a reference. -
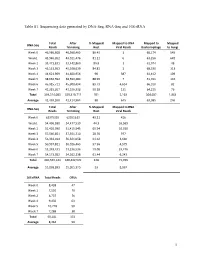
1 Table S1. Sequencing Data Generated by DNA-Seq, RNA-Seq and 16S Rrna
Table S1. Sequencing data generated by DNA-Seq, RNA-Seq and 16S rRNA Total After % Mapped Mapped to DNA Mapped to Mapped DNA-Seq Reads Trimming Host Viral Reads Bacteriophage to Fungi Week 0 46,586,608 46,568,465 86.43 1 68,174 545 Week1 43,940,202 43,911,476 81.11 0 63,256 649 Week 2 33,471,831 33,442,860 89.8 1 61,974 48 Week 3 45,131,953 45,108,639 84.81 2 68,330 313 Week 4 44,621,969 44,600,956 90 387 61,412 109 Week 5 38,630,592 38,590,484 88.99 7 51,011 142 Week 6 45,915,721 45,900,494 89.73 4,634 66,310 82 Week 7 41,215,207 41,196,338 90.18 131 64,215 76 Total 339,514,083 339,319,712 701 5,163 504,682 1,964 Average 42,439,260 42,414,964 88 645 63,085 246 Total After % Mapped Mapped to RNA RNA-Seq Reads Trimming Host Viral Reads Week 0 62375955 62351619 49.21 416 Week1 54,496,930 54,477,530 44.3 16,569 Week 2 51,426,090 51,415,545 65.54 10,016 Week 3 57,566,851 57,551,710 28.76 977 Week 4 56,334,014 56,321,058 61.62 3,660 Week 5 50,937,821 50,926,463 57.56 4,579 Week 6 53,248,431 53,236,536 59.08 29,476 Week 7 54,173,052 54,162,138 61.44 6,243 Total 440,559,144 440,442,599 428 71,936 Average 55,069,893 55,055,325 53 8,992 16S rRNA Total Reads OTUs Week 0 8,438 47 Week 1 7,550 79 Week 2 6,737 76 Week 4 9,450 63 Week 5 10,718 50 Week 7 7,288 38 Total 50,181 353 Average 8,364 59 1 Table S2. -

Proposed Minimal Standards for Describing New Genera and Species of the Suborder Micrococcineae
International Journal of Systematic and Evolutionary Microbiology (2009), 59, 1823–1849 DOI 10.1099/ijs.0.012971-0 Proposed minimal standards for describing new genera and species of the suborder Micrococcineae Peter Schumann,1 Peter Ka¨mpfer,2 Hans-Ju¨rgen Busse 3 and Lyudmila I. Evtushenko4 for the Subcommittee on the Taxonomy of the Suborder Micrococcineae of the International Committee on Systematics of Prokaryotes Correspondence 1DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Inhoffenstraße 7B, P. Schumann 38124 Braunschweig, Germany [email protected] 2Institut fu¨r Angewandte Mikrobiologie, Justus-Liebig-Universita¨t, 35392 Giessen, Germany 3Institut fu¨r Bakteriologie, Mykologie und Hygiene, Veterina¨rmedizinische Universita¨t, A-1210 Wien, Austria 4All-Russian Collection of Microorganisms (VKM), G. K. Skryabin Institute of Biochemistry and Physiology of Microorganisms, RAS, Pushchino, Moscow Region 142290, Russia The Subcommittee on the Taxonomy of the Suborder Micrococcineae of the International Committee on Systematics of Prokaryotes has agreed on minimal standards for describing new genera and species of the suborder Micrococcineae. The minimal standards are intended to provide bacteriologists involved in the taxonomy of members of the suborder Micrococcineae with a set of essential requirements for the description of new taxa. In addition to sequence data comparisons of 16S rRNA genes or other appropriate conservative genes, phenotypic and genotypic criteria are compiled which are considered essential for the comprehensive characterization of new members of the suborder Micrococcineae. Additional features are recommended for the characterization and differentiation of genera and species with validly published names. INTRODUCTION Aureobacterium and Rothia/Stomatococcus) and one pair of homotypic synonyms (Pseudoclavibacter/Zimmer- The suborder Micrococcineae was established by mannella) (Table 1 and http://www.the-icsp.org/taxa/ Stackebrandt et al.