On the Mechanism of Dehydroquinate Synthase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Microbial Production of Methyl Anthranilate, a Grape Flavor Compound
Microbial production of methyl anthranilate, a grape flavor compound Zi Wei Luoa,b,1, Jae Sung Choa,b,1, and Sang Yup Leea,b,c,d,2 aMetabolic and Biomolecular Engineering National Research Laboratory, Department of Chemical and Biomolecular Engineering (BK21 Plus Program), Institute for the BioCentury, Korea Advanced Institute of Science and Technology, 34141 Daejeon, Republic of Korea; bSystems Metabolic Engineering and Systems Healthcare Cross-Generation Collaborative Laboratory, Korea Advanced Institute of Science and Technology, 34141 Daejeon, Republic of Korea; cBioProcess Engineering Research Center, Korea Advanced Institute of Science and Technology, 34141 Daejeon, Republic of Korea; and dBioInformatics Research Center, Korea Advanced Institute of Science and Technology, 34141 Daejeon, Republic of Korea Contributed by Sang Yup Lee, April 5, 2019 (sent for review March 6, 2019; reviewed by Jay D. Keasling and Blaine A. Pfeifer) Methyl anthranilate (MANT) is a widely used compound to give While these biotransformation procedures are considered more grape scent and flavor, but is currently produced by petroleum-based natural and ecofriendly compared with chemical synthesis, their processes. Here, we report the direct fermentative production of actual use is limited due to low yields, long reaction times, and MANT from glucose by metabolically engineered Escherichia coli and formation of byproducts (5). In addition, the chemical and bio- Corynebacterium glutamicum strains harboring a synthetic plant- transformation processes mentioned -

Davidge JBC Supplementary.Pdf
promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/7923/ (includes links to Main Article, Supplementary Material and Figures) Published paper Davidge, K.S., Sanguinetti, G., Yee, C.H., Cox, A.G., McLeod, C.W., Monk, C.E., Mann, B.E., Motterlini, R. and Poole, R.K. (2009) Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators. Journal of Biological Chemistry, 284 (7). pp. 4516-4524. http://dx.doi.org/10.1074/jbc.M808210200 Supplementary Material White Rose Research Online [email protected] Supplementary Material Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators Kelly S Davidge, Guido Sanguinetti, Chu Hoi Yee, Alan G Cox, Cameron W McLeod, Claire E Monk, Brian E Mann, Roberto Motterlini and Robert K Poole Contents Page Number Supplementary Figure S1 3 Inhibition by CORM-3 of E. coli cultures grown in defined medium anaerobically and aerobically Supplementary Figure S2 4 Viability assays showing survival of anaerobically and aerobically E. coli in defined growth medium Supplementary Figure S3 5 Reaction of terminal oxidases in vivo on addition of RuCl2(DMSO)4 to intact cells in a dual-wavelength spectrophotometer Supplementary Figure S4 6 CORM-3 generates carbonmonoxycytochrome bd in vivo and depresses synthesis of cytochrome bo' Supplementary Figure S5 7 Expression of spy-lacZ activity -

Insensitive 3-Deoxy-D-Arabino-Heptulosonate 7-Phosphate Synthaset LISA M
JOURNAL OF BACTERIOLOGY, Nov. 1990, p. 6581-6584 Vol. 172, No. 11 0021-9193/90/116581-04$02.00/0 Copyright X 1990, American Society for Microbiology Cloning of an aroF Allele Encoding a Tyrosine- Insensitive 3-Deoxy-D-arabino-Heptulosonate 7-Phosphate Synthaset LISA M. WEAVER AND KLAUS M. HERRMANN* Department ofBiochemistry, Purdue University, West Lafayette, Indiana 47907 Received 9 May 1990/Accepted 10 August 1990 In Escherichia coli, genes aroF+, aroG+, and aroH+ encode isoenzymes of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthases that are feedback inhibited by tyrosine, phenylalanine, and tryptophan, respectively. A single base pair change in aroF causes a Pro-148-to-Leu-148 substitution and results in a tyrosine-insensitive enzyme. In bacteria and plants, the aromatic amino acids phenyl- plasmid, designated pLW22, contained an 8-kb insert that alanine, tyrosine, and tryptophan are synthesized via the hybridized to the 714-bp DdeI (Fig. 1) aroF probe (6, 11). shikimate pathway (7, 13). The first enzyme of this pathway Digestion of pLW22 with HindIII-BglII gave a 950-bp frag- is 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) ment that hybridized to the probe. This result was unex- synthase (EC 4.1.2.15). In Escherichia coli, the three un- pected, since the corresponding wild-type fragment is 1.8 kb linked genes aroF+, aroG+, and aroH+ (1) encode three in size. Subsequent detailed restriction analysis and hybrid- isoenzymes of DAHP synthase that are sensitive to tyrosine, ization of pLW22 with the 796-bp DdeI (Fig. 1) aroF probe phenylalanine, and tryptophan, respectively (2). -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Advancing a Systems Cell-Free Metabolic Engineering Approach to Natural Product Synthesis and Discovery
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2020 Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery Benjamin Mohr University of Tennessee Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Mohr, Benjamin, "Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery. " PhD diss., University of Tennessee, 2020. https://trace.tennessee.edu/utk_graddiss/6837 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Benjamin Mohr entitled "Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Energy Science and Engineering. Mitchel Doktycz, Major Professor We have read this dissertation and recommend its acceptance: Jennifer Morrell-Falvey, Dale Pelletier, Michael Simpson, Robert Hettich Accepted for the Council: Dixie L. Thompson Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Benjamin Pintz Mohr December 2019 c by Benjamin Pintz Mohr, 2019 All Rights Reserved. -

8.2 Shikimic Acid Pathway
CHAPTER 8 © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FORAromatic SALE OR DISTRIBUTION and NOT FOR SALE OR DISTRIBUTION Phenolic Compounds © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION CHAPTER OUTLINE Overview Synthesis and Properties of Polyketides 8.1 8.5 Synthesis of Chalcones © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC 8.2 Shikimic Acid Pathway Synthesis of Flavanones and Derivatives NOT FOR SALE ORPhenylalanine DISTRIBUTION and Tyrosine Synthesis NOT FOR SALESynthesis OR DISTRIBUTION and Properties of Flavones Tryptophan Synthesis Synthesis and Properties of Anthocyanidins Synthesis and Properties of Isofl avonoids Phenylpropanoid Pathway 8.3 Examples of Other Plant Polyketide Synthases Synthesis of Trans-Cinnamic Acid Synthesis and Activity of Coumarins Lignin Synthesis Polymerization© Jonesof Monolignols & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC Genetic EngineeringNOT FOR of Lignin SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION Natural Products Derived from the 8.4 Phenylpropanoid Pathway Natural Products from Monolignols © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION 119 © Jones & Bartlett Learning, LLC. -

Metabolic Engineering for Improving Anthranilate Synthesis from Glucose
Microbial Cell Factories BioMed Central Research Open Access Metabolic engineering for improving anthranilate synthesis from glucose in Escherichia coli Víctor E Balderas-Hernández1, Andrea Sabido-Ramos1, Patricia Silva1, Natividad Cabrera-Valladares1, Georgina Hernández-Chávez1, José L Báez- Viveros2, Alfredo Martínez1, Francisco Bolívar1 and Guillermo Gosset*1 Address: 1Departamento de Ingeniería Celular y Biocatálisis, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Apdo Postal 510-3, Cuernavaca, Morelos, CP 62210, México and 2Centro de Investigación en Biotecnología, Universidad Autónoma del Estado de Morelos, Av Universidad 2000, Cuernavaca, Morelos, CP 62210, México Email: Víctor E Balderas-Hernández - [email protected]; Andrea Sabido-Ramos - [email protected]; Patricia Silva - [email protected]; Natividad Cabrera-Valladares - [email protected]; Georgina Hernández-Chávez - [email protected]; José L Báez-Viveros - [email protected]; Alfredo Martínez - [email protected]; Francisco Bolívar - [email protected]; Guillermo Gosset* - [email protected] * Corresponding author Published: 2 April 2009 Received: 21 January 2009 Accepted: 2 April 2009 Microbial Cell Factories 2009, 8:19 doi:10.1186/1475-2859-8-19 This article is available from: http://www.microbialcellfactories.com/content/8/1/19 © 2009 Balderas-Hernández et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Anthranilate is an aromatic amine used industrially as an intermediate for the synthesis of dyes, perfumes, pharmaceuticals and other classes of products. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

Suppression of Chorismate Synthase, Which Is Localized in Chloroplasts
www.nature.com/scientificreports OPEN Suppression of chorismate synthase, which is localized in chloroplasts and peroxisomes, results in abnormal fower development and anthocyanin reduction in petunia Shiwei Zhong1,2, Zeyu Chen1, Jinyi Han1, Huina Zhao3, Juanxu Liu1 & Yixun Yu 1,2* In plants, the shikimate pathway generally occurs in plastids and leads to the biosynthesis of aromatic amino acids. Chorismate synthase (CS) catalyses the last step of the conversion of 5-enolpyruvylshikimate 3-phosphate (EPSP) to chorismate, but the role of CS in the metabolism of higher plants has not been reported. In this study, we found that PhCS, which is encoded by a single- copy gene in petunia (Petunia hybrida), contains N-terminal plastidic transit peptides and peroxisomal targeting signals. Green fuorescent protein (GFP) fusion protein assays revealed that PhCS was localized in chloroplasts and, unexpectedly, in peroxisomes. Petunia plants with reduced PhCS activity were generated through virus-induced gene silencing and further characterized. PhCS silencing resulted in reduced CS activity, severe growth retardation, abnormal fower and leaf development and reduced levels of folate and pigments, including chlorophylls, carotenoids and anthocyanins. A widely targeted metabolomics analysis showed that most primary and secondary metabolites were signifcantly changed in pTRV2-PhCS-treated corollas. Overall, the results revealed a clear connection between primary and specialized metabolism related to the shikimate pathway in petunia. Te shikimate pathway -
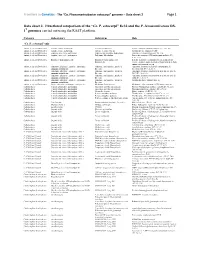
Ca. P. Ectocarpi” Ec32 and the P
Frontiers in Genetics - The “Ca. Phaeomarinobacter ectocarpi” genome – Data sheet 2 Page 1 Data sheet 2. Functional comparison of the “Ca. P. ectocarpi” Ec32 and the P. lavamentivorans DS- 1T genomes carried out using the RAST platform. Category Subcategory Subsystem Role “Ca. P. ectocarpi” only Amino Acids and Derivatives Alanine, serine, and glycine Alanine biosynthesis Valine--pyruvate aminotransferase (EC 2.6.1.66) Amino Acids and Derivatives Alanine, serine, and glycine Glycine cleavage system Sodium/glycine symporter GlyP Amino Acids and Derivatives Arginine; urea cycle, polyamines Arginine and Ornithine Degradation Ornithine cyclodeaminase (EC 4.3.1.12) Amino Acids and Derivatives Arginine; urea cycle, polyamines Polyamine Metabolism Putrescine transport ATP-binding protein PotA (TC 3.A.1.11.1) Amino Acids and Derivatives Branched-chain amino acids Branched-Chain Amino Acid Leucine-responsive regulatory protein, regulator for Biosynthesis leucine (or lrp) regulon and high-affinity branched-chain amino acid transport system Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate periplasmic binding protein ammonia assimilation Bacteria precursor GltI (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate transport system permease protein ammonia assimilation Bacteria GltJ (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate -

Influence of Amino Acid Feeding on Production of Calcimycin
International Journal of Environmental Research and Public Health Article Influence of Amino Acid Feeding on Production of Calcimycin and Analogs in Streptomyces chartreusis Kirstin I. Arend and Julia E. Bandow * Applied Microbiology, Faculty of Biology and Biotechnology, Ruhr University Bochum, 44780 Bochum, Germany; [email protected] * Correspondence: [email protected]; Tel.:+49-234-3223102 Abstract: Streptomyces chartreusis NRRL 3882 produces the polyether ionophore calcimycin and a variety of analogs, which originate from the same biosynthetic gene cluster. The role of calcimycin and its analogs for the producer is unknown, but calcimycin has strong antibacterial activity. Feeding experiments were performed in chemically defined medium systematically supplemented with proteinogenic amino acids to analyze their individual effects on calcimycin synthesis. In the cul- ture supernatants, in addition to known calcimycin analogs, eight so far unknown analogs were detected using LC-MS/MS. Under most conditions cezomycin was the compound produced in highest amounts. The highest production of calcimycin was detected upon feeding with glutamine. Supplementation of the medium with glutamic acid resulted in a decrease in calcimycin production, and supplementation of other amino acids such as tryptophan, lysine, and valine resulted in the decrease in the synthesis of calcimycin and of the known intermediates of the biosynthetic pathway. We demonstrated that the production of calcimycin and its analogs is strongly dependent on amino acid supply. Utilization of amino acids as precursors and as nitrogen sources seem to critically influence calcimycin synthesis. Even amino acids not serving as direct precursors resulted in a Citation: Arend, K.I.; Bandow, J.E. different product profile regarding the stoichiometry of calcimycin analogs. -

Allosteric Regulation of 3 Deoxy-D-Arabino
Allosteric regulation of 3 deoxy-D-arabino- heptulosonate 7-phosphate synthase A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy in Biochemistry at the University of Canterbury by Yifei Fan 2017 Abstract Allostery refers to the process in which interaction of an effector ligand with one site of the protein changes the function of the protein at a distant site. Despite the critical role of allostery in regulation of metabolic pathways, little is known about the details of the allosteric networks and the remarkable diversity in allosteric mechanisms. This study utilises several examples of allosteric proteins to illustrate the interwoven relationships between various allosteric mechanisms, ranging from large conformational changes to subtle dynamic communications. An important metabolic enzyme, 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase (DAH7PS), was selected for this study due to its unique diversity of allosteric regulations and its important role as the first committed step in aromatic amino acid biosynthesis. Essential in many pathogenic bacteria, however lacking mammalian counterparts, DAH7PS and related pathway enzymes provide opportunities in development of novel antimicrobial drugs. The first part of this study addresses the determinants of allosteric ligand selectivity and potency for DAH7PS enzymes that exhibit large conformational changes, and provides structural and functional insights that contribute to the understanding of the role of conformational change in allostery. The second part of this study addresses interchangeability between two different allosteric mechanisms by demonstrating the ease of gene fusion to link two contemporary protein domains and produce functional chimera. The third part of this study addresses the allosteric regulation in DAH7PS enzymes from a different subfamily, for which no large conformational changes are involved in delivering allosteric communication.