Study Protocol, Receive the Treatment in the Established Time Period, and Continue During the Follow-Up Interval
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Time-Updated, Parsimonious Model to Predict AKI in Hospitalized Children
CLINICAL RESEARCH www.jasn.org A Time-Updated, Parsimonious Model to Predict AKI in Hospitalized Children Ibrahim Sandokji ,1,2 Yu Yamamoto ,2 Aditya Biswas,2 Tanima Arora ,2 Ugochukwu Ugwuowo,2 Michael Simonov,2 Ishan Saran,2 Melissa Martin ,2 Jeffrey M. Testani,3 Sherry Mansour,2 Dennis G. Moledina ,2 Jason H. Greenberg ,1,2 and F. Perry Wilson 2 1Department of Pediatrics, Section of Nephrology, Yale University School of Medicine, New Haven, Connecticut 2Clinical and Translational Research Accelerator, Department of Medicine, Yale University School of Medicine, New Haven, Connecticut 3Section of Cardiovascular Medicine, Department of Internal Medicine, Yale University School of Medicine, New Haven, Connecticut ABSTRACT Background Timely prediction of AKI in children can allow for targeted interventions, but the wealth of data in the electronic health record poses unique modeling challenges. Methods We retrospectively reviewed the electronic medical records of all children younger than 18 years old who had at least two creatinine values measured during a hospital admission from January 2014 through January 2018. We divided the study population into derivation, and internal and external valida- tion cohorts, and used five feature selection techniques to select 10 of 720 potentially predictive variables from the electronic health records. Model performance was assessed by the area under the receiver operating characteristic curve in the validation cohorts. The primary outcome was development of AKI (per the Kidney Disease Improving Global Outcomes creatinine definition) within a moving 48-hour win- dow. Secondary outcomes included severe AKI (stage 2 or 3), inpatient mortality, and length of stay. Results Among 8473 encounters studied, AKI occurred in 516 (10.2%), 207 (9%), and 27 (2.5%) encounters in the derivation, and internal and external validation cohorts, respectively. -
D40. Cerebrospinal Fluid.Pdf
CEREBROSPINAL FLUID D40 (1) Cerebrospinal Fluid (CSF) Last updated: June 3, 2019 PHYSIOLOGY ............................................................................................................................................ 1 CSF PRODUCTION .................................................................................................................................. 1 CSF REABSORPTION ............................................................................................................................... 1 PARAMETERS ........................................................................................................................................... 2 NORMAL ................................................................................................................................................ 2 OPENING PRESSURE ................................................................................................................................ 3 COLOR ................................................................................................................................................... 3 BLOODY CSF ......................................................................................................................................... 3 VISCOSITY & TURBIDITY ....................................................................................................................... 4 CELLS ................................................................................................................................................... -

Collected and Prepared By
Body fluids Collected and prepared by Ibtisam H. AlAswad Yousif M. EL-Argan 2017- 2018 1 Table of contents Lab Title Page Introduction 3 Urinary system 5 Lab 1 Urinalysis Specimen collection 10 Physical examination 12 Lab 2 Urinalysis Chemical examination 19 Lab 3 Urineanlysis Microscopic examination 30 Lab 4 Renal calculi 42 Lab 5 Cerebrospinal Macroscopic examination 50 fluid.. Microscopic examination 52 Lab 6 Cerebrospinal Biochemical examination 55 fluid.. Lab 7 Synovial fluid.. Macroscopic examination 61 Microscopic examination 63 Lab 8 Synovial fluid.. Biochemical examination 66 Lab 9 Seminal fluid.. Macroscopic examination 73 Microscopic examination 74 Lab 10 Seminal fluid.. Biochemical examination 78 Lab 11 Serous fluids.. Pleural 84 Lab 12 Serous fluids.. Pericardial 91 Peritoneal 93 Lab 13 Sweat test 97 Sputum analysis 102 Lab 14 Amniotic fluid 105 2 General Introduction The fluids in our bodies are composed of water and dissolved substances, including electrolytes, which are crucial for body function. Movement of Fluids Through the Body We ingest water and electrolytes through the gastrointestinal or GI tract. 1. Absorption. These fluids are absorbed into the plasma in the intestine. 2. Circulation. The fluids circulate within the plasma, bathing the cells in the body. 3. Excretion. The kidneys remove excess ions and water from the body through the urine, although water is also lost at other sites, which will be described later. Water function • Water helps maintain body temperature. When water vaporizes off the skin, it takes large quantities of heat with it. This process cools the body temperature down. • Water acts as a protective cushion in amniotic fluid and cerebrospinal fluid. -

(SPIA) Terminology and Codes Requesting Pathology
Standards for Pathology Informatics in Australia (SPIA) Terminology and Codes Requesting Pathology (v3.0) Superseding and incorporating the Australian Pathology Units and Terminology Standards and Guidelines (APUTS) ISBN: Pending State Health Publication Number (SHPN): Pending Online copyright © RCPA 2017 This work (Standards and Guidelines) is copyright. You may download, display, print and reproduce the Standards and Guidelines for your personal, non- commercial use or use within your organisation subject to the following terms and conditions: 1. The Standards and Guidelines may not be copied, reproduced, communicated or displayed, in whole or in part, for profit or commercial gain. 2. Any copy, reproduction or communication must include this RCPA copyright notice in full. 3. No changes may be made to the wording of the Standards and Guidelines including commentary, tables or diagrams. Excerpts from the Standards and Guidelines may be used. References and acknowledgments must be maintained in any reproduction or copy in full or part of the Standards and Guidelines. Apart from any use as permitted under the Copyright Act 1968 or as set out above, all other rights are reserved. Requests and inquiries concerning reproduction and rights should be addressed to RCPA, 207 Albion St, Surry Hills, NSW 2010, Australia. This material contains content from LOINC® (http://loinc.org). The LOINC table, LOINC codes, LOINC panels and forms file, LOINC linguistic variants file, LOINC/RSNA Radiology Playbook, and LOINC/IEEE Medical Device Code Mapping Table are copyright © 1995-2016, Regenstrief Institute, Inc. and the Logical Observation Identifiers Names and Codes (LOINC) Committee and is available at no cost under the license at http://loinc.org/terms-of-use.” This material includes SNOMED Clinical Terms® (SNOMED CT®) which is used by permission of the International Health Terminology Standards Development Organisation (IHTSDO®). -

Laboratory Tests Recommendations Table and Guidelines For
Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (DMD) Common Data Elements (CDEs) The Laboratory Tests CDE recommendations have been divided into separate categories and sorted alphabetically. The recommendations are presented in different tables as shown below: • Table 1 – CBC with Differential • Table 2 – Chemistries • Table 3 – CSF • Table 4 – CSF Culture • Table 5 – Blood Tests • Table 6 – Blood Gas Tests • Table 7 – Urinalysis • Table 8 – Scalp Electroencephalogram • Table 9 – Immune Cell Subsets The tables provide an extensive list of the possible laboratory tests that may be collected in a study. While this list is broad, it is not exhaustive. The Laboratory Tests have all been recommended as supplemental or exploratory as the type of laboratory tests collected is dependent on the nature of the study and therefore none of the recommendations are considered core. The list should be used in conjunction with the Laboratory Tests and Tracking Case Report Form (CRF). Version 1.0 Page 1 of 15 Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (DMD) Common Data Elements (CDEs) Table 1. CBC with Differential CDE General Comments Purpose Classification Absolute Basophils Measured in % Safety Supplemental Absolute Eosinophils Measured in % Safety Supplemental Absolute Lymphocytes Measured in % Safety Supplemental Absolute Monocytes Measured in % Safety Supplemental Absolute Neutrophils Measured in % Safety Supplemental Basophils Measured in cells per microliter or % Safety Supplemental Blasts Measured in cells -
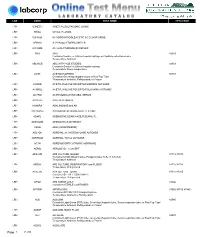
Lab Code Test Name Cpt4 Code
LAB CODE TEST NAME CPT4 CODE LRH U3METH 3-METHYLGLUTACONIC, URINE LRH 5HIAA 5-HIAA, PLASMA LRH U5HIA24 5-HYDROXYINDOLEACETIC ACID 24HR URINE LRH APHAG A. PHAGOCYTOPHILUM PCR LRH A1CGMK A1c w/GLYCOMARK(R) REFLEX LRH ABG ABG 82803 Container:Sodium or Lithium heparin syringe or Capillary collection device Temperature:Ambient LRH ABGHGB ABG WITH HGB STUDIES 82803 Container:Sodium or Lithium heparin syringe Temperature:Room temperature LRH ACET ACETAMINOPHEN 80307 Container:Green-top (heparin) tube or Red-Top Tube Temperature:Ambient, Refrigerated, or Frozen LRH ACHRB ACETYLCHOLINE RECEPTOR BINDING AUTOABS LRH ACHRBL ACETYLCHOLINE RECEPTOR BLOCKING AUTOABS LRH ACHRM AChR MODULATING ABS, SERUM LRH ADAPER ADA, PERITONEAL LRH HUMIRA ADALIMUMAB and AB LRH ADENOAG ADENO/ROTO VIRUS AG DETECTION LRH ADAPL ADENOSINE DEAMINASE,PLEURAL FL LRH ADENOAB ADENOVIRUS ANTIBODY LRH VASO ADH (VASOPRESSIN) LRH AD21OH ADRENAL 21-HYDROXYLASE AUTOABS LRH ADRENAB ADRENAL TOTAL AUTOABS LRH ACTH ADRENOCORTICOTROPIC HORMONE LRH AERID AEROBE ID + SUSCEPT LRH AFBUVB AFB CULTURE, BLOOD 87116 87015 Container:UVMC Blood Culture/Fungal Isolater Tube (1.5-10mL) Temperature:Ambient LRH AFBUV AFB CULTURE, RESPIRATORY and FLUIDS 87116 87015 Temperature:Refrigerated LRH AFBUVU AFB CULTURE, URINE 87116 87015 Container:STERILE CONTAINER Temperature:Refrigerated LRH AFSM AFB SMEAR ONLY 87206 Container:STERILE CONTAINER LRH AFFIRM AFFIRM VPIII 87660 87510 87480 Container:AFFIRM VPIII transport system Temperature:Ambient or Refrigerated LRH ALB ALBUMIN 82040 Container:Light Green (PST) -

Effects of Blood Contamination on Cerebrospinal Fluid Cell Counts, Protein, and D-Dimer Concentrations John Henry Rossmeisl, Jr
Effects of Blood Contamination on Cerebrospinal Fluid Cell Counts, Protein, and D-dimer Concentrations John Henry Rossmeisl, Jr. Thesis submitted to the Faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of Master of Science In Veterinary Medical Science Gregory C. Troy, Co-Chair G. Daniel Boon, Co-ChairÑ Karen Dyer Inzana Bernard Jortner Ñ Deceased January 5, 2003, Blacksburg, Virginia Keywords: Cerebrospinal Fluid, Blood, D-dimers, Dog, Central Nervous System Copyright 2003, John Henry Rossmeisl, Jr. Effects of Blood Contamination on Cerebrospinal Fluid Cell Counts, Protein, and D-dimer Concentrations John Henry Rossmeisl, Jr. (Abstract) Cerebrospinal fluid analysis (CSF) is commonly performed in clinical neurology, and is a sensitive, but non-specific indicator of central nervous system (CNS) pathology. Blood contaminated CSF samples have the potential to adversely affect results of cytologic, serologic, microbiologic, and molecular biologic diagnostics. A clear consensus of the effects of blood contamination on CSF analysis could not be drawn following a review of the existing veterinary literature. Based on data from earlier reports, it was hypothesized that iatrogenic blood contamination of CSF would result in significant increases in both the CSF total protein (TP) concentration and nucleated cell count (WBC). As hypothesized, in vitro CSF blood contamination resulted in statistically significant (p < 0.01) linear increases in both the CSF TP and WBC with increasing RBC concentration in CSF from sixteen normal dogs. Although increases in TP and WBC are statistically significant, their clinical impact is negligible. Results of this study demonstrate that in normal dogs, the mean CSF TP concentration collected from the cerebellomedullary cistern, is lower than previously reported. -

APUTS) Terminology and Codes Requesting Pathology (V2.1
AUSTRALIAN PATHOLOGY UNITS AND TERMINOLOGY (APUTS) Terminology and Codes Requesting Pathology (v2.1) ISBN: Pending 1 State Health Publication Number (SHPN): Pending Online copyright © RCPA 2014 This work (Standards and Guidelines) is copyright. You may download, display, print and reproduce the Standards and Guidelines for your personal, non- commercial use or use within your organisation subject to the following terms and conditions: 1. The Standards and Guidelines may not be copied, reproduced, communicated or displayed, in whole or in part, for profit or commercial gain. 2. Any copy, reproduction or communication must include this RCPA copyright notice in full. 3. No changes may be made to the wording of the Standards and Guidelines including commentary, tables or diagrams. Excerpts from the Standards and Guidelines may be used. References and acknowledgments must be maintained in any reproduction or copy in full or part of the Standards and Guidelines. Apart from any use as permitted under the Copyright Act 1968 or as set out above, all other rights are reserved. Requests and inquiries concerning reproduction and rights should be addressed to RCPA, 207 Albion St, Surry Hills, NSW 2010, Australia. This material contains content from LOINC® (http://loinc.org). The LOINC table, LOINC codes and LOINC panels and forms file are copyright © 1995-2012, Regenstrief Institute, Inc. and the Logical Observation Identifiers Names and Codes (LOINC) Committee and available at no cost under the license athttp://loinc.org/terms-of-use. This material includes SNOMED Clinical Terms® (SNOMED CT®) which is used by permission of the International Health Terminology Standards Development Organisation (IHTSDO®). -

The Biochemistry of Body Fluids
The Biochemistry of Body Fluids Association of Clinical Biochemists in Ireland Alan Balfe Mark Kilbane Stan Barry Peadar McGing Ophelia Blake Ruth O’Kelly Dermot Cannon Paula O’Shea Martin Healy Editors: Dr Peadar McGing, Ms Ruth O’Kelly Guest Editor: Dr Yvonne O’Meara, Consultant Nephrologist The Asssociation of Clinical Biochemists in Ireland - The Biochemistry of Body Fluids Contents Produced by the Scientific Committee of the Preface V Association of Clinical Biochemists in Ireland (ACBI) Introduction VII Cerebral Spinal Fluid 1 Pleural Fluid 11 First edition: October 2009 Pericardial Fluid 15 Ascitic / Peritoneal Fluid 17 Sweat 21 Amniotic Fluid 25 Saliva 29 Seminal Fluid 33 Synovial Fluid 35 Other booklets in this series Guidelines on the use of biochemical cardiac markers and risk factors Guidelines on the use of therapeutic drug monitoring Guidelines for the use of tumour markers Version 1.0 October 2009 V The Asssociation of Clinical Biochemists in Ireland - The Biochemistry of Body Fluids The Asssociation of Clinical Biochemists in Ireland - The Biochemistry of Body Fluids Preface These Guidelines are the latest in a series commissioned and produced by the Scientific Committee of the Association of Clinical Biochemists in Ireland, to promote appropriate and effective use of the laboratory service. They are intended to be a concise reference document to assist practitioners in the Clinical Biochemistry field and those who order tests from the laboratory service, whether hospital- or community-based. This volume deals with the analyses of body fluids other than blood and urine. It covers analytical issues, interpretation of results and limitations of testing. -

Biochemical Markers of Time Since Death In
Biochemical markers of time since death in cerebrospinal fluid: A first step towards “Forensomics” Pierre-Antoine Peyron, Sylvain Lehmann, Constance Delaby, Eric Baccino, Christophe Hirtz To cite this version: Pierre-Antoine Peyron, Sylvain Lehmann, Constance Delaby, Eric Baccino, Christophe Hirtz. Bio- chemical markers of time since death in cerebrospinal fluid: A first step towards “Forensomics”. Critical Reviews in Clinical Laboratory Sciences, Taylor & Francis, 2019, 56 (4), pp.274-286. 10.1080/10408363.2019.1619158. hal-02505396 HAL Id: hal-02505396 https://hal.archives-ouvertes.fr/hal-02505396 Submitted on 11 Mar 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Critical Reviews in Clinical Laboratory Sciences ISSN: 1040-8363 (Print) 1549-781X (Online) Journal homepage: https://www.tandfonline.com/loi/ilab20 Biochemical markers of time since death in cerebrospinal fluid: a first step towards “Forensomics” Pierre-Antoine Peyron, Sylvain Lehmann, Constance Delaby, Eric Baccino & Christophe Hirtz To cite this article: Pierre-Antoine Peyron, Sylvain Lehmann, Constance Delaby, Eric Baccino & Christophe Hirtz (2019): Biochemical markers of time since death in cerebrospinal fluid: a first step towards “Forensomics”, Critical Reviews in Clinical Laboratory Sciences, DOI: 10.1080/10408363.2019.1619158 To link to this article: https://doi.org/10.1080/10408363.2019.1619158 Accepted author version posted online: 15 May 2019. -

Elevated Cerebrospinal Fluid Protein in POLG‐Related Epilepsy
Accepted: 24 May 2018 DOI: 10.1111/epi.14459 FULL-LENGTH ORIGINAL RESEARCH Elevated cerebrospinal fluid protein in POLG-related epilepsy: Diagnostic and prognostic implications Omar Hikmat1,2 | Karin Naess3,4 | Martin Engvall3,5 | Claus Klingenberg6,7 | Magnhild Rasmussen8,9 | Chantal M. E. Tallaksen10,11 | Eylert Brodtkorb12,13 | Torunn Fiskerstrand14,15 | Pirjo Isohanni16,17 | Johanna Uusimaa18,19 | Niklas Darin20 | Shamima Rahman21,22 | Laurence A. Bindoff2,23 1Department of Pediatrics, Haukeland University Hospital, Bergen, Norway 2Department of Clinical Medicine (K1), University of Bergen, Bergen, Norway 3Center for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm, Sweden 4Department of Medical Biochemistry and Biophysics, Karolinska Institute, Stockholm, Sweden 5Department of Molecular Medicine and Surgery, Karolinska Institute, Stockholm, Sweden 6Department of Pediatric and Adolescent Medicine, University Hospital of North Norway, Tromso, Norway 7Pediatric Research Group, Department of Clinical Medicine, UiT–Arctic University of Norway, Tromso, Norway 8Women and Children’s Division, Department of Clinical Neurosciences for Children, Oslo University Hospital, Oslo, Norway 9Unit for Congenital and Hereditary Neuromuscular Disorders, Department of Neurology, Oslo University Hospital, Oslo, Norway 10Department of Neurology, Oslo University Hospital, Oslo, Norway 11Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway 12Department of Neuroscience, Norwegian University of Science