THESIS for DOCTORAL DEGREE (Ph.D.)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Limited Presence of IL-22 Binding Protein, a Natural IL-22 Inhibitor, Strengthens Psoriatic Skin Inflammation
Limited Presence of IL-22 Binding Protein, a Natural IL-22 Inhibitor, Strengthens Psoriatic Skin Inflammation This information is current as Jérôme C. Martin, Kerstin Wolk, Gaëlle Bériou, Ahmed of September 25, 2021. Abidi, Ellen Witte-Händel, Cédric Louvet, Georgios Kokolakis, Lucile Drujont, Laure Dumoutier, Jean-Christophe Renauld, Robert Sabat and Régis Josien J Immunol 2017; 198:3671-3678; Prepublished online 29 March 2017; Downloaded from doi: 10.4049/jimmunol.1700021 http://www.jimmunol.org/content/198/9/3671 References This article cites 47 articles, 12 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/198/9/3671.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 25, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Limited Presence of IL-22 Binding Protein, a Natural IL-22 Inhibitor, Strengthens Psoriatic Skin Inflammation Je´roˆme C. -

Supplementary Material
Coagulation & Its Disorders SUPPLEMENTARY APPENDIX Dexamethasone promotes durable factor VIII-specific tolerance in hemophilia A mice via thymic mechanisms Maria T. Georgescu, 1 Paul C. Moorehead, 2,3 Alice S. van Velzen, 4 Kate Nesbitt, 1 Birgit M. Reipert, 5 Katharina N. Steinitz, 5 Maria Schuster, 5 Christine Hough 1 and David Lillicrap 1 1Department of Pathology and Molecular Medicine, Queen’s University, Kingston, ON, Canada; 2Janeway Children’s Health and Reha - bilitation Centre, St. John’s, NL, Canada; 3Faculty of Medicine, Memorial University, St. John’s, NL, Canada; 4Department of Pediatric Hematology, Immunology and Infectious Diseases, Emma Children’s Hospital, Amsterdam, the Netherlands and 5Baxalta Innovations GmbH, Vienna, Austria ©2018 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2018.189852 Received: January 30, 2018. Accepted: April 19, 2018. Pre-published: April 19, 2018. Correspondence: [email protected] Supplementary Material 100 80 60 40 20 Anti-VWF IgG Positive IgG Mice (%)Anti-VWF 0 FVIII/FVIII FVIII+Dex/FVIII FVIII+Dex/intFVIII+FVIII n=4 n=13 n=12 Figure S1. Administration of Dex during initial FVIII exposure does not impair the immune response to VWF. Anti-VWF IgG incidence at week 22, following exposure to VWF (Week 18-21) in mice initially treated with FVIII or FVIII+Dex and with no evidence of anti-FVIII IgG at week 4. Table S1. Genes down-regulated following FVIII+Dex treatment. Transcript Count Ratio Gene Name FVIII+Dex vs FVIII Dex vs HBSS Ccr4 -5.82 -14.45 Rag1 -4.18 -16.00 Cd69 -3.51 -4.40 Ccr9 -3.46 -9.90 Slamf1 -3.20 -6.50 Cd8b1 -3.18 -7.77 Cd40lg -2.99 -2.56 Cxcl1 -2.90 -2.37 Rag2 -2.84 -9.21 Rorc -2.78 -5.65 Cd8a -2.67 -7.74 Cd4 -2.66 -5.06 Il9 -2.64 -2.26 Il12b -2.60 -2.49 Bcl6 -2.59 -3.30 Il27 -2.54 -3.06 Mr1 -2.48 -3.26 Sh2d1a -2.39 -6.68 Il13 -2.23 -2.38 Ccl22 -2.22 -2.51 Il16 -2.22 -3.05 Socs1 -2.20 -4.55 Card9 -2.12 -3.32 Cxcr4 -2.12 -3.98 Tcf7 -2.12 -5.16 Lck -2.06 -4.47 Icam2 -2.02 -3.09 Table S2. -

IL-22 Binding Protein Promotes the Disease Process in Multiple Sclerosis Hannes Lindahl, André O
IL-22 Binding Protein Promotes the Disease Process in Multiple Sclerosis Hannes Lindahl, André O. Guerreiro-Cacais, Sahl Khalid Bedri, Mathias Linnerbauer, Magdalena Lindén, Nada This information is current as Abdelmagid, Karolina Tandre, Claire Hollins, Lorraine of September 24, 2021. Irving, Colin Glover, Clare Jones, Lars Alfredsson, Lars Rönnblom, Ingrid Kockum, Mohsen Khademi, Maja Jagodic and Tomas Olsson J Immunol published online 10 July 2019 Downloaded from http://www.jimmunol.org/content/early/2019/07/09/jimmun ol.1900400 Supplementary http://www.jimmunol.org/content/suppl/2019/07/10/jimmunol.190040 http://www.jimmunol.org/ Material 0.DCSupplemental Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 24, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2019 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published -

System, Method and Software for Calculation of a Cannabis Drug Efficiency Index for the Reduction of Inflammation
International Journal of Molecular Sciences Article System, Method and Software for Calculation of a Cannabis Drug Efficiency Index for the Reduction of Inflammation Nicolas Borisov 1,† , Yaroslav Ilnytskyy 2,3,†, Boseon Byeon 2,3,4,†, Olga Kovalchuk 2,3 and Igor Kovalchuk 2,3,* 1 Moscow Institute of Physics and Technology, 9 Institutsky lane, Dolgoprudny, Moscow Region 141701, Russia; [email protected] 2 Department of Biological Sciences, University of Lethbridge, Lethbridge, AB T1K 3M4, Canada; [email protected] (Y.I.); [email protected] (B.B.); [email protected] (O.K.) 3 Pathway Rx., 16 Sandstone Rd. S., Lethbridge, AB T1K 7X8, Canada 4 Biomedical and Health Informatics, Computer Science Department, State University of New York, 2 S Clinton St, Syracuse, NY 13202, USA * Correspondence: [email protected] † First three authors contributed equally to this research. Abstract: There are many varieties of Cannabis sativa that differ from each other by composition of cannabinoids, terpenes and other molecules. The medicinal properties of these cultivars are often very different, with some being more efficient than others. This report describes the development of a method and software for the analysis of the efficiency of various cannabis extracts to detect the anti-inflammatory properties of the various cannabis extracts. The method uses high-throughput gene expression profiling data but can potentially use other omics data as well. According to the signaling pathway topology, the gene expression profiles are convoluted into the signaling pathway activities using a signaling pathway impact analysis (SPIA) method. The method was tested by inducing inflammation in human 3D epithelial tissues, including intestine, oral and skin, and then exposing these tissues to various extracts and then performing transcriptome analysis. -

Low Interleukin-22 Binding Protein Is Associated with High Mortality In
ARTICLE 1 Low Interleukin-22 Binding Protein Is Associated With High Mortality in Alcoholic Hepatitis and Modulates LIVER Interleukin-22 Receptor Expression 08/26/2020 on BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3PE2KhmxLsUKwRUb9LEtaRQ9wXW4bkT7EyguzqSnjJYw= by https://journals.lww.com/ctg from Downloaded Sidsel Støy, MD, PhD1, Tea Lund Laursen, MD1, Emilie Glavind, MD, PhD1, Peter Lykke Eriksen, MD, PhD1, Ewa Terczynska-Dyla, PhD2, Downloaded Nils Erik Magnusson, PhD3, Stephen Hamilton-Dutoit, MD, PhD4, Frank Viborg Mortensen, MD, PhD5, Sanne Skovgård Veidal, PhD6, Kristoffer Rigbolt, PhD6, Oliviero Riggio, MD, PhD7, Bent Deleuran, MD, DMSc8, Hendrik Vilstrup, MD, DSc1 and from Thomas Damgaard. Sandahl, MD, PhD1 https://journals.lww.com/ctg INTRODUCTION: In alcoholic hepatitis (AH), high interleukin (IL)-22 production is associated with disease improvement, purportedly through enhanced infection resistance and liver regeneration. IL-22 binding protein (BP) by binds and antagonizes IL-22 bioactivity, but data on IL-22BP in liver disease suggest a complex BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3PE2KhmxLsUKwRUb9LEtaRQ9wXW4bkT7EyguzqSnjJYw= interplay. Despite the scarcity of human data, IL-22 is in clinical trial as treatment of AH. We, therefore, in patients with AH, described the IL-22 system focusing on IL-22BP and associations with disease course, and mechanistically pursued the human associations in vitro. METHODS: We prospectively studied 41 consecutive patients with AH at diagnosis, days 7 and 90, and followed them for up to 1 year. We measured IL-22 pathway proteins in liver biopsies and blood and investigated IL-22BP effects on IL-22 in hepatocyte cultures. RESULTS: IL-22BP was produced in the gut and was identifiable in the patients with AH’ livers. -

Downloaded Genes (Degs) in Ileum and Lung of Chicken Infected with H5N1 and from "Japanese Quail (Coturnix Japonica) Genome Se- H5N2
Cao et al. BMC Medical Genomics 2017, 10(Suppl 4):70 DOI 10.1186/s12920-017-0304-z RESEARCH Open Access Differential responses of innate immunity triggered by different subtypes of influenza a viruses in human and avian hosts Yingying Cao1, Yaowei Huang1,KeXu1, Yuanhua Liu1, Xuan Li2,YeXu3*, Wu Zhong4* and Pei Hao1* From 16th International Conference on Bioinformatics (InCoB 2017) Shenzhen, China. 20-22 September 2017 Abstract Background: Innate immunity provides first line of defense against viral infections. The interactions between hosts and influenza A virus and the response of host innate immunity to viral infection are critical determinants for the pathogenicity or virulence of influenza A viruses. This study was designed to investigate global changes of gene expression and detailed responses of innate immune systems in human and avian hosts during the course of infection with various subtypes of influenza A viruses, using collected and self-generated transcriptome sequencing data from human bronchial epithelial (HBE), human tracheobronchial epithelial (HTBE), and A549 cells infected with influenza A virus subtypes, namely H1N1, H3N2, H5N1 HALo mutant, and H7N9, and from ileum and lung of chicken and quail infected with H5N1, or H5N2. Results: We examined the induction of various cytokines and chemokines in human hosts infected with different subtypes of influenza A viruses. Type I and III interferons were found to be differentially induced with each subtype. H3N2 caused abrupt and the strongest response of IFN-β and IFN-λ, followed by H1N1 (though much weaker), whereas H5N1 HALo mutant and H7N9 induced very minor change in expression of type I and III interferons. -

Xo GENE PANEL
xO GENE PANEL Targeted panel of 1714 genes | Tumor DNA Coverage: 500x | RNA reads: 50 million Onco-seq panel includes clinically relevant genes and a wide array of biologically relevant genes Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 -

Supporting Information
Supporting Information Voigt et al. 10.1073/pnas.1705165114 SI Materials and Methods the HTCR includes obtaining written informed consent from all Mice. BALB/c mice were purchased from Janvier. C57BL/6 mice patients with lung cancer and has been approved by the Ethics were purchased from Charles River. Mice were 5 wk old at the Committee of the Medical Faculty, LMU Munich (no. 025-12) and − − onset of experiments. Spleens from IL-1R / mice were provided by the Bavarian State Medical Association. All operations of from T. Stöger, Helmholtz Center Munich, Munich, and spleens Biobank are certified according to ISO 9001:2008 (57). Written − − from IL-23 / mice were a gift from K. Savvatis, Charité, Berlin. informed consent was obtained from all patients with breast cancer before collection of specimens, in line with the respective in- Cell Lines. The murine lung cancer cell line 1 (Line-1) was kindly stitutional policies and in accordance with to the Declaration of provided by Nejat K. Egilmez, University of Louisville, Louisville, Helsinki. Tumor specimens were obtained from patients un- KY (55), and cells were cultured in complete RPMI 1640 medium dergoing clinically indicated surgery. Histological analysis of (Lonza). If not stated otherwise, growth media used were supple- NSCLC or breast cancer was made by a pathologist as part of mented with 10% FBS, 100 μg/mL streptomycin, 1 IU/mL peni- standard of care. Ethical approval was obtained from the Ethics cillin, and 2 mM L-glutamine (Life Technologies). The 4T1murine Committee of the Medical Faculty, LMU Munich (reference breast cancer cell line was kindly provided by Maria Wartenberg, nos. -

KRAS Mutations Are Negatively Correlated with Immunity in Colon Cancer
www.aging-us.com AGING 2021, Vol. 13, No. 1 Research Paper KRAS mutations are negatively correlated with immunity in colon cancer Xiaorui Fu1,2,*, Xinyi Wang1,2,*, Jinzhong Duanmu1, Taiyuan Li1, Qunguang Jiang1 1Department of Gastrointestinal Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People's Republic of China 2Queen Mary College, Medical Department, Nanchang University, Nanchang, Jiangxi, People's Republic of China *Equal contribution Correspondence to: Qunguang Jiang; email: [email protected] Keywords: KRAS mutations, immunity, colon cancer, tumor-infiltrating immune cells, inflammation Received: March 27, 2020 Accepted: October 8, 2020 Published: November 26, 2020 Copyright: © 2020 Fu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The heterogeneity of colon cancer tumors suggests that therapeutics targeting specific molecules may be effective in only a few patients. It is therefore necessary to explore gene mutations in colon cancer. In this study, we obtained colon cancer samples from The Cancer Genome Atlas, and the International Cancer Genome Consortium. We evaluated the landscape of somatic mutations in colon cancer and found that KRAS mutations, particularly rs121913529, were frequent and had prognostic value. Using ESTIMATE analysis, we observed that the KRAS-mutated group had higher tumor purity, lower immune score, and lower stromal score than the wild- type group. Through single-sample Gene Set Enrichment Analysis and Gene Set Enrichment Analysis, we found that KRAS mutations negatively correlated with enrichment levels of tumor infiltrating lymphocytes, inflammation, and cytolytic activities. -

Human Colon Cancer Primer Library
Human Colon Cancer Primer Library Catalog No: HCCR-1 Supplier: RealTimePrimers Lot No: XXXXX Supplied as: solid Stability: store at -20°C Description Contains 88 primer sets directed against cytokine and chemokine receptor genes and 8 housekeeping gene primer sets. Provided in a 96-well microplate (20 ul - 10 uM). Perform up to 100 PCR arrays (based on 20 ul assay volume per reaction). Just add cDNA template and SYBR green master mix. Gene List: • CCR1 chemokine (C-C motif) receptor 1 • IL11RA interleukin 11 receptor, alpha • CCR2 chemokine (C-C motif) receptor 2 • IL11RB interleukin 11 receptor, beta • CCR3 chemokine (C-C motif) receptor 3 • IL12RB1 interleukin 12 receptor, beta 1 • CCR4 chemokine (C-C motif) receptor 4 • IL12RB2 interleukin 12 receptor, beta 2 • CCR5 chemokine (C-C motif) receptor 5 • IL13RA1 interleukin 13 receptor, alpha 1 • CCR6 chemokine (C-C motif) receptor 6 • IL13RA2 interleukin 13 receptor, alpha 2 • CCR7 chemokine (C-C motif) receptor 7 • IL15RA interleukin 15 receptor, alpha • CCR8 chemokine (C-C motif) receptor 8 • IL15RB interleukin 15 receptor, beta • CCR9 chemokine (C-C motif) receptor 9 • IL17RA interleukin 17 receptor A • CCR10 chemokine (C-C motif) receptor 10 • IL17RB interleukin 17 receptor B • CX3CR1 chemokine (C-X3-C motif) receptor 1 • IL17RC interleukin 17 receptor C • CXCR1 chemokine (C-X-C motif) receptor 1 • IL17RD interleukin 17 receptor D • CXCR2 chemokine (C-X-C motif) receptor 2 • IL17RE interleukin 17 receptor E • CXCR3 chemokine (C-X-C motif) receptor 3 • IL18R1 interleukin 18 receptor -

Human Inflammation Panel a Gene Set You Can Count On
PRODUCT BULLETIN nCounter® Human Inflammation Panel A Gene Set You Can Count On The nCounter Human Inflammation Panel is a comprehensive assay of 249 human genes known to be differentially expressed in inflammation. The gene list represents a broad range of inflammation-related pathways, including: • Chemokine • Integrin signaling • Cytokine • Oxidative stress response • Interleukin • B cell activation • Toll receptor • T cell activiation This gene list was compiled by querying several public databases for inflammation-related genes. Each gene was verified to be differentially expressed under conditions leading to inflammation. Verification was performed using MSigDB, a repository of gene expression data developed by researchers at the Massachusetts Institute of Technology and the Broad Institute1. Other public databases were used to obtain functional gene expression information for each gene. The final nCounter Human Inflammation Panel consists of 249 inflammation-related genes and six internal reference genes. For the gene list and additional information about this panel, visit the nCounter Pre-built Panels product page at nanostring.com. Product Highlights nCounter Analysis System Overview • Simple No need for cross-referencing databases The nCounter Analysis System from NanoString offers a cost- effective way to easily profile hundreds of gene transcripts • Highly Curated simultaneously with high sensitivity and precision. The digital Our expert bioinformaticists use a very detection of target molecules and high levels of multiplexing rigorous process in selecting the most eliminate the compromise between data quality and data meaningful set of genes quantity, bringing better sensitivity, reproducibility, and linearity to your results. It is ideal for studying defined gene sets across • Efficient a large sample set, e.g., microarray validation, pathway analysis, Multiplexed assay profiles 249 human biomarker validation, and splice variation analysis. -
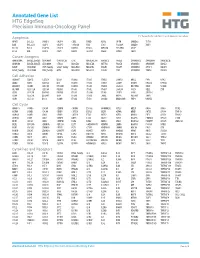
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1