Analysis and Interpretation of Microarray Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pathways and Networks Biological Meaning of the Gene Sets
Pathways and Networks Biological meaning of the gene sets Gene ontology terms ? Pathway mapping Linking to Pubmed abstracts or associted MESH terms Regulation by the same transcription factor (module) Protein families and domains Gene set enrichment analysis Over representation analysis 1 Gene set enrichment analysis 1. Given an a priori defined set of genes S. 2. Rank genes (e.g. by t‐value between 2 groups of microarray samples) ranked gene list L. 3. Calculation of an enrichment score (ES) that reflects the degree to which a set S is overrepresented at the extremes (top or bottom) of the entire ranked list L. 4. Estimation the statistical significance (nominal P value) of the ES by using an empirical phenotype‐based permutation test procedure. 5. Adjustment for multiple hypothesis testing by controlling the false discovery rate (FDR). Gene set enrichment analysis Subramanian A et al. Proc Natl Acad Sci (2005) 2 Biochemical and Metabolic Pathways Böhringer Mannheim Signaling networks (in cancer cell) Hannah, Weinberg, Cell. 2000 137 NCI curated pathway maps (http://pid.nci.nih.gov) Signal Transduction Knowledge Environment http://stke.sciencemag.org/cm/ 3 Pathways • Pathways are available mechanistic information •Kyoto Encylopedia of Genes and Genomes, KEGG (http://www.genome.jp/kegg/) BioCYc • EcoCYC Metabolic Pathway Database (E. Coli) •Also for other organisms (e.g. HumanCYC) 4 Pathways • Pathways from Biocarta • http://www.biocarta.com/genes/index.asp Transpath Part of larger BioBase package (commercial) • PathwayBuilder package -
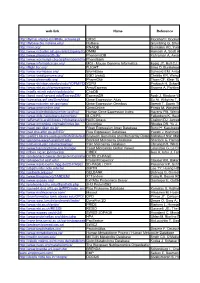
Web-Link Name Reference DRSC Flockhart I, Booker M, Kiger A, Et Al.: Flyrnai: the Drosophila Rnai Screening Center Database
web-link Name Reference http://flyrnai.org/cgi-bin/RNAi_screens.pl DRSC Flockhart I, Booker M, Kiger A, et al.: FlyRNAi: the Drosophila RNAi screening center database. Nucleic Acids Res. 34(Database issue):D489-94 (2006) http://flybase.bio.indiana.edu/ FlyBase Grumbling G, Strelets V.: FlyBase: anatomical data, images and queries. Nucleic Acids Res. 34(Database issue):D484-8 (2006) http://rnai.org/ RNAiDB Gunsalus KC, Yueh WC, MacMenamin P, et al.: RNAiDB and PhenoBlast: web tools for genome-wide phenotypic mapping projects. Nucleic Acids Res. 32(Database issue):D406-10 (2004) http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIMOMIM Hamosh A, Scott AF, Amberger JS, et al.: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33(Database issue):D514-7 (2005) http://www.phenomicdb.de/ PhenomicDB Kahraman A, Avramov A, Nashev LG, et al.: PhenomicDB: a multi-species genotype/phenotype database for comparative phenomics. Bioinformatics 21(3):418-20 (2005) http://www.worm.mpi-cbg.de/phenobank2/cgi-bin/MenuPage.pyPhenoBank http://www.informatics.jax.org/ MGI - Mouse Genome Informatics Eppig JT, Bult CJ, Kadin JA, et al.: The Mouse Genome Database (MGD): from genes to mice--a community resource for mouse biology. Nucleic Acids Res. 33(Database issue):D471-5 (2005) http://flight.licr.org/ FLIGHT Sims D, Bursteinas B, Gao Q, et al.: FLIGHT: database and tools for the integration and cross-correlation of large-scale RNAi phenotypic datasets. Nucleic Acids Res. 34(Database issue):D479-83 (2006) http://www.wormbase.org/ WormBase Schwarz EM, Antoshechkin I, Bastiani C, et al.: WormBase: better software, richer content. -

Genmapp 2: New Features and Resources for Pathway Analysis Nathan Salomonis Gladstone Institute of Cardiovascular Disease
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2007 GenMAPP 2: New features and resources for pathway analysis Nathan Salomonis Gladstone Institute of Cardiovascular Disease Kristina Hanspers Gladstone Institute of Cardiovascular Disease Alexander C. Zambon Gladstone Institute of Cardiovascular Disease Karen Vranizan Gladstone Institute of Cardiovascular Disease Steven C. Lawlor Gladstone Institute of Cardiovascular Disease See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Part of the Medicine and Health Sciences Commons Recommended Citation Salomonis, Nathan; Hanspers, Kristina; Zambon, Alexander C.; Vranizan, Karen; Lawlor, Steven C.; Dahlquist, Kam D.; Doniger, Scott .;W Stuart, Josh; Conklin, Bruce R.; and Pico, Alexander R., ,"GenMAPP 2: New features and resources for pathway analysis." BMC Bioinformatics.,. 217. (2007). https://digitalcommons.wustl.edu/open_access_pubs/208 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Nathan Salomonis, Kristina Hanspers, Alexander C. Zambon, Karen Vranizan, Steven C. Lawlor, Kam D. Dahlquist, Scott .W Doniger, Josh Stuart, Bruce R. Conklin, and Alexander R. Pico This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/208 -

Genmapp 2: New Features and Resources for Pathway Analysis Nathan Salomonis Gladstone Institute of Cardiovascular Disease
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2007 GenMAPP 2: New features and resources for pathway analysis Nathan Salomonis Gladstone Institute of Cardiovascular Disease Kristina Hanspers Gladstone Institute of Cardiovascular Disease Alexander C. Zambon Gladstone Institute of Cardiovascular Disease Karen Vranizan Gladstone Institute of Cardiovascular Disease Steven C. Lawlor Gladstone Institute of Cardiovascular Disease See next page for additional authors Follow this and additional works at: http://digitalcommons.wustl.edu/open_access_pubs Part of the Medicine and Health Sciences Commons Recommended Citation Salomonis, Nathan; Hanspers, Kristina; Zambon, Alexander C.; Vranizan, Karen; Lawlor, Steven C.; Dahlquist, Kam D.; Doniger, Scott .;W Stuart, Josh; Conklin, Bruce R.; and Pico, Alexander R., ,"GenMAPP 2: New features and resources for pathway analysis." BMC Bioinformatics.8,. 217. (2007). http://digitalcommons.wustl.edu/open_access_pubs/208 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Nathan Salomonis, Kristina Hanspers, Alexander C. Zambon, Karen Vranizan, Steven C. Lawlor, Kam D. Dahlquist, Scott .W Doniger, Josh Stuart, Bruce R. Conklin, and Alexander R. Pico This open access publication is available at Digital Commons@Becker: http://digitalcommons.wustl.edu/open_access_pubs/208 -

In-Cell Architecture of an Actively Transcribing-Translating Expressome
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.28.970111; this version posted February 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. In-cell architecture of an actively transcribing-translating expressome Francis J. O’Reilly1, †, Liang Xue2,3, †, Andrea Graziadei1, †, Ludwig Sinn1, Swantje Lenz1, 5 Dimitry Tegunov4, Cedric Blötz5, Wim J. H. Hagen2, Patrick Cramer4, Jörg Stülke5, Julia Mahamid2,*, Juri Rappsilber1,6,* Affiliations: 1 Bioanalytics, Institute of Biotechnology, Technische Universität Berlin, 13355 Berlin, 10 Germany 2 Structural and Computational Biology Unit, European Molecular Biology Laboratory (EMBL), Meyerhofstraße 1, 69117 Heidelberg, Germany. 3 Collaboration for joint PhD degree between EMBL and Heidelberg University, Faculty of Biosciences 15 4 Department of Molecular Biology, Max-Planck-Institute for Biophysical Chemistry, Am Faßberg 11, 37077, Göttingen, Germany 5 Department of General Microbiology, Institute of Microbiology and Genetics, GZMB, Georg- August-University Göttingen, Grisebachstraße 8, 37077 Göttingen, Germany 6 Wellcome Centre for Cell Biology, University of Edinburgh, Max Born Crescent, Edinburgh, 20 EH9 3BF, UK *Correspondence to: [email protected], [email protected] †These authors contributed equally to this work. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.28.970111; this version posted February 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -

Expressomal Approach for Comprehensive Analysis and Visualization of Ligand Sensitivities of Xenoestrogen Responsive Genes
Expressomal approach for comprehensive analysis and visualization of ligand sensitivities of xenoestrogen responsive genes Toshi Shiodaa,b,1, Noël F. Rosenthala, Kathryn R. Cosera, Mizuki Sutoa, Mukta Phatakc, Mario Medvedovicc, Vincent J. Careyb,d, and Kurt J. Isselbachera,b,1 aMolecular Profiling Laboratory, Massachusetts General Hospital Center for Cancer Research, Charlestown, MA 02129; bDepartment of Medicine, Harvard Medical School, Boston, MA 02115; cLaboratory for Statistical Genomics and Systems Biology, Department of Environmental Health, University of Cincinnati College of Medicine, Cincinnati, OH 45267; and dChanning Laboratory, Brigham and Women’s Hospital, Boston, MA 02115 Contributed by Kurt J. Isselbacher, August 26, 2013 (sent for review June 17, 2013) Although biological effects of endocrine disrupting chemicals Evidence is accumulating that the EDCs may cause significant (EDCs) are often observed at unexpectedly low doses with occa- biological effects in humans or animals at doses far lower than sional nonmonotonic dose–response characteristics, transcriptome- the exposure limits set by regulatory agencies (8, 9). In addition wide profiles of sensitivities or dose-dependent behaviors of the to such low-dose effects, an increasing number of studies also EDC responsive genes have remained unexplored. Here, we describe support the concept of the nonmonotonic EDC effects, whose dose–response curves show U shapes or inverted-U shapes (8- expressome analysis for the comprehensive examination of dose- – dependent gene responses -

Bioinformaticsbioinformatics Introduction to Genomics and Proteomics I
www. .uni-rostock.de BioinformaticsBioinformatics Introduction to genomics and proteomics I Ulf Schmitz [email protected] Bioinformatics and Systems Biology Group www.sbi.informatik.uni-rostock.de Ulf Schmitz, Introduction to genomics and proteomics I 1 www. .uni-rostock.de Outline Genomics/Genetics 1. The tree of life • Prokaryotic Genomes –Bacteria – Archaea • Eukaryotic Genomes – Homo sapiens 2. Genes • Expression Data Ulf Schmitz, Introduction to genomics and proteomics I 2 www. .uni-rostock.de Genomics - Definitions #Genetics: is the science of genes, heredity, and the variation of organisms. Humans began applying knowledge of genetics in prehistory with the domestication and breeding of plants and animals. In modern research, genetics provides tools in the investigation of the function of a particular gene, e.g. analysis of genetic interactions. #Genomics: attempts the study of large-scale genetic patterns across the genome for a given species. It deals with the systematic use of genome information to provide answers in biology, medicine, and industry. Genomics has the potential of offering new therapeutic methods for the treatment of some diseases, as well as new diagnostic methods. Major tools and methods related to genomics are bioinformatics, genetic analysis, measurement of gene expression, and determination of gene function. Ulf Schmitz, Introduction to genomics and proteomics I 3 Genes www. .uni-rostock.de •a gene coding for a protein corresponds to a sequence of nucleotides along one or more regions of a molecule of DNA • in species with double stranded DNA (dsDNA), genes may appear on either strand • bacterial genes are continuous regions of DNA bacterium: • a string of 3N nucleotides encodes a string of N amino acids • or a string of N nucleotides encodes a structural RNA molecule of N residues eukaryote: • a gene may appear split into separated segments in the DNA • an exon is a stretch of DNA retained in mRNA that the ribosomes translate into protein Ulf Schmitz, Introduction to genomics and proteomics I 4 www. -

1471-2105-8-217.Pdf
BMC Bioinformatics BioMed Central Software Open Access GenMAPP 2: new features and resources for pathway analysis Nathan Salomonis1,2, Kristina Hanspers1, Alexander C Zambon1, Karen Vranizan1,3, Steven C Lawlor1, Kam D Dahlquist4, Scott W Doniger5, Josh Stuart6, Bruce R Conklin1,2,7,8 and Alexander R Pico*1 Address: 1Gladstone Institute of Cardiovascular Disease, 1650 Owens Street, San Francisco, CA 94158 USA, 2Pharmaceutical Sciences and Pharmacogenomics Graduate Program, University of California, 513 Parnassus Avenue, San Francisco, CA 94143, USA, 3Functional Genomics Laboratory, University of California, Berkeley, CA 94720 USA, 4Department of Biology, Loyola Marymount University, 1 LMU Drive, MS 8220, Los Angeles, CA 90045 USA, 5Computational Biology Graduate Program, Washington University School of Medicine, St. Louis, MO 63108 USA, 6Department of Biomolecular Engineering, University of California, Santa Cruz, CA 95064 USA, 7Department of Medicine, University of California, San Francisco, CA 94143 USA and 8Department of Molecular and Cellular Pharmacology, University of California, San Francisco, CA 94143 USA Email: Nathan Salomonis - [email protected]; Kristina Hanspers - [email protected]; Alexander C Zambon - [email protected]; Karen Vranizan - [email protected]; Steven C Lawlor - [email protected]; Kam D Dahlquist - [email protected]; Scott W Doniger - [email protected]; Josh Stuart - [email protected]; Bruce R Conklin - [email protected]; Alexander R Pico* - [email protected] * Corresponding author Published: 24 June 2007 Received: 16 November 2006 Accepted: 24 June 2007 BMC Bioinformatics 2007, 8:217 doi:10.1186/1471-2105-8-217 This article is available from: http://www.biomedcentral.com/1471-2105/8/217 © 2007 Salomonis et al; licensee BioMed Central Ltd. -

Structural Basis of Mitochondrial Translation Shintaro Aibara1†, Vivek Singh1,2, Angelika Modelska3‡, Alexey Amunts1,2*
RESEARCH ARTICLE Structural basis of mitochondrial translation Shintaro Aibara1†, Vivek Singh1,2, Angelika Modelska3‡, Alexey Amunts1,2* 1Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Solna, Sweden; 2Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden; 3Laboratory of Translational Genomics, Centre for Integrative Biology, University of Trento, Trento, Italy Abstract Translation of mitochondrial messenger RNA (mt-mRNA) is performed by distinct mitoribosomes comprising at least 36 mitochondria-specific proteins. How these mitoribosomal proteins assist in the binding of mt-mRNA and to what extent they are involved in the translocation of transfer RNA (mt-tRNA) is unclear. To visualize the process of translation in human mitochondria, we report ~3.0 A˚ resolution structure of the human mitoribosome, including the L7/L12 stalk, and eight structures of its functional complexes with mt-mRNA, mt-tRNAs, recycling factor and additional trans factors. The study reveals a transacting protein module LRPPRC-SLIRP that delivers mt-mRNA to the mitoribosomal small subunit through a dedicated platform formed by the mitochondria-specific protein mS39. Mitoribosomal proteins of the large subunit mL40, mL48, and *For correspondence: mL64 coordinate translocation of mt-tRNA. The comparison between those structures shows [email protected] dynamic interactions between the mitoribosome and its ligands, suggesting a sequential Present address: †Department mechanism of conformational changes. of Molecular Biology, Max- Planck-Institute for BiophysicalChemistry, Go¨ ttingen, Germany; ‡Aix Marseille Introduction Universite´, CNRS, INSERM, Translation in humans takes place in the cytosol and mitochondria. Mitochondrial translation is Centre d’Immunologie responsible for the maintenance of the cellular energetic balance through synthesis of proteins deMarseille-Luminy (CIML), Marseille, France involved in oxidative phosphorylation. -

Mycobacterial Held Is a Nucleic Acids-Clearing Factor for RNA
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.20.211821; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Mycobacterial HelD is a nucleic acids-clearing factor for RNA 2 polymerase 3 4 5 Tomáš Koubaa*#, Tomáš Koval’b*, Petra Sudzinovác*, Jiří Pospíšilc, Barbora Brezovskác, Jarmila 6 Hnilicovác, Hana Šanderovác, Martina Janouškovác, Michaela Šikovác, Petr Haladac, Michal 7 Sýkorad, Ivan Barvíke, Jiří Nováčekf, Mária Trundováb, Jarmila Duškováb, Tereza Skálováb, 8 URee Chong, Katsuhiko S. Murakamig, Jan Dohnálekb#, Libor Krásnýc# 9 10 a EMBL Grenoble, 71 Avenue des Martyrs, France 11 b Institute of Biotechnology of the Czech Academy of Sciences, Centre BIOCEV, Prumyslova 12 595, 25250 Vestec, Czech Republic 13 c Institute of Microbiology of the Czech Academy of Sciences, Prague, Czech Republic 14 d Institute of Molecular Genetics of the Czech Academy of Sciences, Prague, Czech Republic 15 e Charles University, Faculty of Mathematics and Physics, Institute of Physics, Prague, Czech 16 Republic 17 f CEITEC, Masaryk University, Brno, Czech Republic 18 g Department of Biochemistry and Molecular Biology, The Center for RNA Molecular Biology, 19 Pennsylvania State University, University Park, PA 16802, USA 20 21 22 23 24 25 *Equal contribution 26 #Corresponding author: [email protected], [email protected], [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.20.211821; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Chromatin Remodelling Complex Dosage Modulates Transcription Factor Function in Heart Development
ARTICLE Received 5 Aug 2010 | Accepted 11 Jan 2011 | Published 8 Feb 2011 DOI: 10.1038/ncomms1187 Chromatin remodelling complex dosage modulates transcription factor function in heart development Jun K. Takeuchi1,2,*, Xin Lou3,*, Jeffrey M. Alexander1,4, Hiroe Sugizaki2, Paul Delgado-Olguín1, Alisha K. Holloway1, Alessandro D. Mori1, John N. Wylie1, Chantilly Munson5,6, Yonghong Zhu3, Yu-Qing Zhou7, Ru-Fang Yeh8, R. Mark Henkelman7,9, Richard P. Harvey10,11, Daniel Metzger12, Pierre Chambon12, Didier Y. R. Stainier4,5,6, Katherine S. Pollard1,8, Ian C. Scott3,13 & Benoit G. Bruneau1,4,5,14 Dominant mutations in cardiac transcription factor genes cause human inherited congenital heart defects (CHDs); however, their molecular basis is not understood. Interactions between transcription factors and the Brg1/Brm-associated factor (BAF) chromatin remodelling complex suggest potential mechanisms; however, the role of BAF complexes in cardiogenesis is not known. In this study, we show that dosage of Brg1 is critical for mouse and zebrafish cardiogenesis. Disrupting the balance between Brg1 and disease-causing cardiac transcription factors, including Tbx5, Tbx20 and Nkx2–5, causes severe cardiac anomalies, revealing an essential allelic balance between Brg1 and these cardiac transcription factor genes. This suggests that the relative levels of transcription factors and BAF complexes are important for heart development, which is supported by reduced occupancy of Brg1 at cardiac gene promoters in Tbx5 haploinsufficient hearts. Our results reveal complex dosage-sensitive interdependence between transcription factors and BAF complexes, providing a potential mechanism underlying transcription factor haploinsufficiency, with implications for multigenic inheritance of CHDs. 1 Gladstone Institute of Cardiovascular Disease, San Francisco, California 94158, USA.