A Highly Sensitive and Selective Spectrofluorimetric Method for The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Arsenic Incorporation Into Garden Vegetables Irrigated with Contaminated Water
JASEM ISSN 1119-8362 Full-text Available Online at J. Appl. Sci. Environ. Manage. December, 2007 All rights reserved www.bioline.org.br/ja Vol. 11(4) 105 - 112 Arsenic Incorporation into Garden Vegetables Irrigated with Contaminated Water RAHMAN I.M.M. * AND HASAN M.T. Applied Research Laboratory, Department of Chemistry, University of Chittagong, Chittagong-4331, Bangladesh *Corresponding Author. Tel.: +88-031-635631. Fax: +88-031-726310. E-mail: [email protected] ABSTRACT: Daily vegetable requirement are mostly fulfilled in Bangladesh through homestead garden production which are usually irrigated with arsenic-rich underground water. Garden vegetables grown in arsenic-tainted soil may uptake and accumulate significant amount of arsenic in their tissue. Mean, minimum and maximum arsenic content in some common garden vegetables, e.g. bean, bitter gourd, bottle gourd, brinjal, chilli, green papaya, mint, okra, palwal, potato, red amaranth, string bean and sweet gourd, from an arsenic- prone locality of Bangladesh have been assessed. The contribution of vegetable-arsenic in the daily diet was estimated. Correlation with the groundwater arsenic status and statistical significance of variations has been discussed. @ JASEM The natural contamination of shallow hand extracted from the shallow tubewells, were tubewells in Bangladesh with arsenic has caused collected in pre-washed polyethylene bottles. widespread human exposure to this toxic element 0.01% HNO3 per litre of water was added as through drinking water (Karim, 2000, Paul et al., preservative and kept at 4°C before analysis. Ag- 2000). Beside direct consumption for drinking, DDTC-Hexamethylenetetramine-Chloroform arsenic contaminated water is also used for method, having detection limit 0.20 µg/mL of irrigation and cooking purposes. -

Bounced Back List.Xlsx
SL Cycle Name Beneficiary Name Bank Name Branch Name Upazila District Division Reason for Bounce Back 1 Jan/21-Jan/21 REHENA BEGUM SONALI BANK LTD. NA Bagerhat Sadar Upazila Bagerhat Khulna 23-FEB-21-R03-No Account/Unable to Locate Account 2 Jan/21-Jan/21 ABDUR RAHAMAN SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number SHEIKH 3 Jan/21-Jan/21 KAZI MOKTADIR HOSEN SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 4 Jan/21-Jan/21 BADSHA MIA SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 5 Jan/21-Jan/21 MADHAB CHANDRA SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number SINGHA 6 Jan/21-Jan/21 ABDUL ALI UKIL SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 7 Jan/21-Jan/21 MRIDULA BISWAS SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 8 Jan/21-Jan/21 MD NASU SHEIKH SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 9 Jan/21-Jan/21 OZIHA PARVIN SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 10 Jan/21-Jan/21 KAZI MOHASHIN SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 11 Jan/21-Jan/21 FAHAM UDDIN SHEIKH SONALI BANK LTD. NA Chitalmari Upazila Bagerhat Khulna 16-FEB-21-R04-Invalid Account Number 12 Jan/21-Jan/21 JAFAR SHEIKH SONALI BANK LTD. -

Sitakunda Upazila Chittagong District Bangladesh
Integrated SMART Survey Nutrition, Care Practices, Food Security and Livelihoods, Water Sanitation and Hygiene Sitakunda Upazila Chittagong District Bangladesh January 2018 Funded By Acknowledgement Action Against Hunger conducted Baseline Integrated SMART Nutrition survey in Sitakunda Upazila in collaboration with Institute of Public Health Nutrition (IPHN). Action Against Hunger would like to acknowledge and express great appreciation to the following organizations, communities and individuals for their contribution and support to carry out SMART survey: District Civil Surgeon and Upazila Health and Family Planning Officer for their assistance for successful implementation of the survey in Sitakunda Upazila. Action Against Hunger-France for provision of emergency response funding to implement the Integrated SMART survey as well as technical support. Leonie Toroitich-van Mil, Health and Nutrition Head of department of Action Against Hunger- Bangladesh for her technical support. Mohammad Lalan Miah, Survey Manager for executing the survey, developing the survey protocol, providing training, guidance and support to the survey teams as well as the data analysis and writing the final survey report. Action Against Hunger Cox’s Bazar for their logistical support and survey financial management. Mothers, Fathers, Caregivers and children who took part in the assessment during data collection. Action Against Hunger would like to acknowledge the community representatives and community people who have actively participated in the survey process for successful completion of the survey. Finally, Action Against Hunger is thankful to all of the surveyors, supervisor and Survey Manager for their tremendous efforts to successfully complete the survey in Sitakunda Upazila. Statement on Copyright © Action Against Hunger | Action Contre la Faim Action Against Hunger (ACF) is a non-governmental, non-political and non-religious organization. -

Tor for Preparation of Development Plan for Mirsharai Upazila, Chittagong District: Risk Sensitive Landuse Plan
Selection of Web Firm for Web Site Designing Development & Hosting ToR for Preparation of Development Plan for Mirsharai Upazila, Chittagong District: Risk Sensitive Landuse Plan ANNEX-X TERMS OF REFERENCE (TOR) for Selection of Web Firm for Web Site Designing Development & Hosting Under ÒPÆMÖvg †Rjvi gximivB Dc‡Rjvi Dbœqb cwiKíbv cÖYqb t mvwe©K `y‡h©vM e¨e¯’vcbv‡K f~wg e¨env‡ii gva¨‡g m¤ú„³KiYÓ (Preparation of Development Plan for Mirsharai Upazila, Chattogram District: Risk Sensitive Landuse Plan) URBAN DEVELOPMENT DIRECTORATE Ministry of Housing and Public Works Government of the People’s Republic of Bangladesh November, 2016 Signature of the Bidder Signature of the Project Director Signature of the Director 131 Selection of Web Firm for Web Site Designing Development & Hosting ToR for Preparation of Development Plan for Mirsharai Upazila, Chittagong District: Risk Sensitive Landuse Plan Table of Content Content Page No. Appendix-01: Background Information of the Project 133 Appendix-02: Scope of Work of the Assignment 136 Appendix-03: Bidding For Tender 141 Appendix-04: Format of Curriculum Vitae and Project Team 142 Signature of the Bidder Signature of the Project Director Signature of the Director 132 Selection of Web Firm for Web Site Designing Development & Hosting ToR for Preparation of Development Plan for Mirsharai Upazila, Chittagong District: Risk Sensitive Landuse Plan APPENDIX 01 BCKGROUND INFORMATION OF THE PROJECT 1.1. Project Background Mirsharai Upazila (CHATTOGRAM DISTRICT) area 482.88 sqkm(BBS)/509.80sqkm(GIS Data), located in between 22°39' and 22°59' north latitudes and in between 91°27' and 91°39' east longitudes. -

Report on AK Taj Group Masrur M. A. Hoque.Pdf (983.4Kb)
Internship Report on AK TAJ GROUP Prepared for, MD. Tamzidul Islam Assistant Professor BRAC BusinessSchool BRAC University Prepared By, Masrur M. A. Hoque ID # 12164092 Submission Date – 15/12/2015 LETTER OF TRANSMITTAL December 15, 2015 MD. Tamzidul Islam Assistant Professor BRAC BusinessSchool BRAC University Subject: Internship Report. Dear Sir, I would like to thank you for supervising and helping me throughout the semester. With due respect I am submitting a copy of intern report foryourappreciation. I have given my best effort to prepare the report with relevant information that I have collected from an onsite production department which is belongs to a group of company and from other sources during my accomplishthe course. I have the immense pleasure to have the opportunity to study on the marketing practices of AK TAJ Group. There is no doubt that the knowledge I have gathered during the study will help me in real life. For your kind consideration I would like to mention that there might be some errors and mistakes due to limitations of my knowledge. I expect that you will forgive me considering that I am still learner and in the process of learning. Thanking for your time and reviews. Yours faithfully Masrur M. A. Hoque ID-12164092 BRAC Business School BRAC University Acknowledgement The successful completion of this internship might not be possible in time without the help some person whose suggestion and inspiration made it happen. First of all I want to thank my Course Instructor MD. Tamzidul Islam for guiding me during the course. Without his help this report would not have been accomplished. -

Human Rights Report 2015 on Indigenous Peoples in Bangladesh
HHUMANUMAN RRIGHTSIGHTS RREPORTEPORT 20152015 R H ONON INDIGENOUSINDIGENOUS PEOPLESPEOPLES EPORT 2015 EPORT About Kapaeeng Foundation UMAN ININ BANGLADESHBANGLADESH Kapaeeng Foundation as a human rights orgnisation for Indigenous Peoples in Bangladesh was established on 1 April 2004 with the view to working for promotion and R protection of the rights of Indigenous Peoples in the IGHTS country. The term ‘Kapaeeng’ is derived from indigenous Khumi language, meaning ‘Rights’. Kapaeeng Foundation is duly registered with the Office of the Registrar of Joint INBANGLADESH ON INDIGENOUS PEOPLES Stock Companies and Firms of Commerce Ministry, Government of Peoples’ Republic of Bangladesh under the Societies Registration Act XXI of 1860. Vision: The vision of Kapaeeng Foundation is to establish a society based on the values of justice, equity and freedom where human rights and fundamental freedoms of Indigenous Peoples are ensured. Mission : The mission of Kapaeeng Foundation are, among others, to promote and protect the human rights of the Indigenous Peoples of the country and to conduct advocacy, lobby and campaign programme for the same at local, national, regional and international level; to establish a strong network and partnership with national, regional and international organizations and individuals working on promotion and protection of human rights as well as Indigenous Peoples rights and to raise the capacity of the Indigenous Peoples in promoting and protecting the human rights. ISBN 978-984-34-0664-4 Human Rights Report 2015 on Indigenous Peoples in Bangladesh First Edition January 2016 Human Rights Report 2015 Copyright © Kapaeeng Foundation on Indigenous Peoples Published by in Bangladesh Kapaeeng Foundation House # 23/25, Salma Garden, Road # 4 PC Culture Housing, Block # B, Mohammadpur, Dhaka-1207 Tel: +88-02-8190801 E-mail: [email protected] Web: www.kapaeeng.org Supported by Oxfam House 4, Road 3, Block I, Banani Dhaka 1213, Bangladesh Editor Tel: +88-02-8813607-9 Prof. -

List of Upazilas of Bangladesh
List Of Upazilas of Bangladesh : Division District Upazila Rajshahi Division Joypurhat District Akkelpur Upazila Rajshahi Division Joypurhat District Joypurhat Sadar Upazila Rajshahi Division Joypurhat District Kalai Upazila Rajshahi Division Joypurhat District Khetlal Upazila Rajshahi Division Joypurhat District Panchbibi Upazila Rajshahi Division Bogra District Adamdighi Upazila Rajshahi Division Bogra District Bogra Sadar Upazila Rajshahi Division Bogra District Dhunat Upazila Rajshahi Division Bogra District Dhupchanchia Upazila Rajshahi Division Bogra District Gabtali Upazila Rajshahi Division Bogra District Kahaloo Upazila Rajshahi Division Bogra District Nandigram Upazila Rajshahi Division Bogra District Sariakandi Upazila Rajshahi Division Bogra District Shajahanpur Upazila Rajshahi Division Bogra District Sherpur Upazila Rajshahi Division Bogra District Shibganj Upazila Rajshahi Division Bogra District Sonatola Upazila Rajshahi Division Naogaon District Atrai Upazila Rajshahi Division Naogaon District Badalgachhi Upazila Rajshahi Division Naogaon District Manda Upazila Rajshahi Division Naogaon District Dhamoirhat Upazila Rajshahi Division Naogaon District Mohadevpur Upazila Rajshahi Division Naogaon District Naogaon Sadar Upazila Rajshahi Division Naogaon District Niamatpur Upazila Rajshahi Division Naogaon District Patnitala Upazila Rajshahi Division Naogaon District Porsha Upazila Rajshahi Division Naogaon District Raninagar Upazila Rajshahi Division Naogaon District Sapahar Upazila Rajshahi Division Natore District Bagatipara -
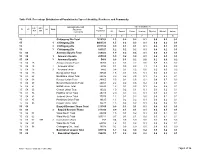
Chittagong C09.Pdf
Table C-09: Percentage Distribution of Population by Type of disability, Residence and Community Administrative Unit Type of disability (%) UN / MZ / Total ZL UZ Vill RMO Residence WA MH Population Community All Speech Vision Hearing Physical Mental Autism 1 2 3 4 5 6 7 8 9 10 15 Chittagong Zila Total 7616352 1.3 0.2 0.2 0.1 0.5 0.2 0.1 15 1 Chittagong Zila 4463723 1.5 0.2 0.3 0.1 0.6 0.2 0.1 15 2 Chittagong Zila 2971102 0.9 0.1 0.1 0.1 0.4 0.1 0.1 15 3 Chittagong Zila 181527 1.2 0.2 0.2 0.1 0.4 0.2 0.1 15 04 Anowara Upazila Total 259022 1.9 0.2 0.4 0.1 0.8 0.3 0.1 15 04 1 Anowara Upazila 253556 1.9 0.2 0.4 0.1 0.8 0.3 0.1 15 04 3 Anowara Upazila 5466 0.8 0.1 0.2 0.0 0.2 0.0 0.2 15 04 15 Anowara Union Total 10260 4.2 0.3 2.2 0.6 0.8 0.2 0.2 15 04 15 1 Anowara Union 4794 8.1 0.5 4.5 1.1 1.5 0.3 0.3 15 04 15 3 Anowara Union 5466 0.8 0.1 0.2 0.0 0.2 0.0 0.2 15 04 19 Bairag Union Total 30545 1.8 0.1 0.5 0.1 0.6 0.3 0.1 15 04 28 Barakhain Union Total 28836 3.0 0.2 0.5 0.1 1.8 0.3 0.1 15 04 38 Barasat Union Total 28865 2.5 0.2 0.6 0.2 0.8 0.7 0.2 15 04 47 Burumchhara Union Total 20061 2.2 0.2 0.6 0.2 1.0 0.1 0.1 15 04 57 Battali Union Total 23630 1.3 0.1 0.2 0.1 0.5 0.2 0.1 15 04 66 Chatari Union Total 19022 1.0 0.2 0.1 0.1 0.3 0.2 0.1 15 04 76 Haildhar Union Total 25315 2.0 0.4 0.3 0.1 0.7 0.3 0.1 15 04 81 Juidandi Union Total 17575 1.2 0.1 0.2 0.0 0.6 0.1 0.1 15 04 85 Paraikora Union Total 19635 1.3 0.2 0.2 0.1 0.6 0.2 0.1 15 04 95 Roypur Union Total 35278 1.5 0.2 0.2 0.1 0.6 0.2 0.1 15 06 Bayejid Bostami Thana Total 211355 0.9 0.1 0.1 0.1 0.4 0.1 0.1 15 06 2 Bayejid Bostami Thana 211355 0.9 0.1 0.1 0.1 0.4 0.1 0.1 15 06 01 Ward No-01 Total 29209 1.5 0.1 0.2 0.1 0.9 0.1 0.1 15 06 02 Ward No-02 Total 103314 0.7 0.1 0.1 0.0 0.3 0.1 0.1 15 06 02 998 2 Cantonment (Part) 758 0.4 0.0 0.0 0.0 0.1 0.0 0.3 15 06 03 Ward No-03 Total 68794 1.2 0.1 0.2 0.1 0.5 0.2 0.1 15 06 98 Chittagang Cnt. -

Geo-Electrical Resistivity Survey for Fresh Groundwater Investigation in Mirsharai Economic Zone, Chittagong in the South-Eastern Coastal Areas of Bangladesh
14 Journal of Nepal Geological Society, 2020, vol. 60, pp. 181-194 https://doi.org/10.3126/jngs.v60i0.31262 Geo-electrical resistivity survey for fresh groundwater investigation in Mirsharai Economic Zone, Chittagong in the south-eastern coastal areas of Bangladesh *A.S.M. Woobaidullah, Md. Ariful Islam, Md. Zakir Hossain, Md. Shahidul Islam Geology Department, Dhaka University, Dhaka 1000, Bangladesh *Corresponding author’s email: [email protected] ABSTRACT Salinity in the groundwater is one of the major concerning issues in the coastal region of Bangladesh. Mirsharai Economic Zone, Mirsharai Upazila in the south-eastern coastal region of Chittagong District of Bangladesh, requires substantial amount of fresh groundwater supply for industrial and household use. The purpose of this study is to delineate the aquifer system of the study area and to determine the extension of potential fresh water aquifer for groundwater development through geophysical electrical resistivity sounding survey. Vertical Electrical Sounding in conjunction with borehole data provides information about the saline-fresh water interface, depth distribution, thickness of the fresh-water aquifers and local lithology. Lithological cross section shows that the sedimentary deposition and aquifer-aquitard distribution of this region are irregular even within a short spatial distance. The surface layer of top soil of clay or silty clay composition identified as aquitard shows resistivity in the ranges from 1.24 Ωm to 11 Ωm. This aquitard is underlain by a sand layer acting as shallow aquifer of varying thickness shows resistivity ranging mostly from 1.9 Ωm to 11Ωm reflecting the pore space water as saline to brackish. -

Environmental Study of Mirsharai Upazila, Chattogram Different
iffi Environmental Study of Mirsharai Upazila, Chattogram Sound, Air Pollution (Dust Particles) and Water Quality in Different Locations U RBAN DEVELOPMENT DIRECTORATE (U DD) Ministry of Housing and Public Works Government of the People's Republic of Bangladesh June, 2019 Environmental Study of Mirsharai Upazila, Chattogram Sound, Air Pollution (Dust Particles) and Water Quality in Different Locations ,*i- URBAN DEVELOPMENT DIRECTORATE (UDD) Ministry of Housing and Public Works Government of the People's Republic of Bangladesh lrne,2019 I ) Acknowledgement This study has been conducted by Urban Deveropment Directorate ur. is gratefur (uDD) assisted by . ,ro of Ministry Qtex sorutions ::ir:H;::1X?, rorr,n, ana puoric "i w",0. assigning ",111 to to a t h o s e o * T:_Til ",. monitoring " ",,I ffi ff [r I Jf and reporting' we wouh ;'j,fl 1''.n*::HtI":iT:: :fi ri'r"," give speciar thr;k;to professor :: : I ;: Biochemistrv Dr. Hossain Uddin :ffi;J:T.i}:of anJ rvroru.rr* Biorogy, or.,r,..'rr,rersity Shekhar for his conrinuous support the team members or etex sorurions Ltd. who has provided the necessary and:!lrHJi:*l1rt"^:" testing,.., ,r,";;;.'b;, ",i#:::TiilruL,.:ff1[ffi::::,ff"lT ," ,n" monitoring ,N I Abbreviations & Acronyrns DoE Department of Environment ECR Environmental Conservation Rule ECA Environmental Conservation Act lFc I nternational Finance Corporation OSHA Occu pationa I Safety a nd Hea lth Ad ml n istration EZ Economic Zone APHA American Public Health Association db Decibel PM Particulate Matter TDS Total Dissolved Solids DO Dissolved Oxygen BOD Biochemical Oxygen Demand coD Chemical Oxygen demand llt' lil I Executive Summary The research work is carried out to get a picture of Noise, Air Pollution and Water quality in different locations of Mirsharai Upazila, Chattogram & to set a baseline environmental standard conducive for im plementi ng the necessa ry projects. -

Simultaneous Determination of Arsenic (III)
s z Available online at http://www.journalcra.com INTERNATIONAL JOURNAL OF CURRENT RESEARCH International Journal of Current Research Vol. 12, Issue, 02, pp.10363-10379, February, 2020 DOI: https://doi.org/10.24941/ijcr.38058.02.2020 ISSN: 0975-833X RESEARCH ARTICLE A SIMPLE, HIGHLY SELECTIVE SPECTROPHOTOMETRIC METHOD FOR THE SIMULTANEOUS DETERMINATION OF ARSENIC (III) & ARSENIC (V)) AT NANO-TRACE LEVELS IN SOME GROUNDWATER, REAL, BIOLOGICAL, FOOD AND SOIL SAMPLES USING 2-HYDROXY-1- NAPTHALDEHYDE-ORTHOAMINOPHENOL 1,*Jamaluddin Ahmed, M., 1,2Afrin, Ayesha and 1 Imran Hossain Mollah, M. 1Laboratory of Analytical Chemistry, Department of Chemistry, University of Chittagong, Chittagong, Bangladesh 2Department of Applied Chemistry and Chemical Engineering, University of Chittagong, Chittagong, Bangladesh ARTICLE INFO ABSTRACT Article History: A new spectrophotometric reagent 2-Hydroxy-1-napthaldehyde-orthoaminophenol has been Received 14th November, 2019 synthesized and characterized. A very simple, ultra-sensitive, highly selective and non-extractive Received in revised form new spectrophotometric method for the simultaneous determination of arsenic (III) and arsenic (V) at 20th December, 2019 nano-trace levels using 2-Hydroxy-1-napthaldehyde-orthoaminophenol (HNA-OAP)has been Accepted 29th January, 2020 developed. HNA-OAP has been proposed as a new analytical reagent for the direct non-extractive th Published online 28 February, 2020 spectrophotometric determination of arsenic (III&V). This novel spectrophotometric reagent reacts in a slightly acidic (0.00016-0.0004 M H2SO4) aqueous solution with arsenic(III) in 20% N,N- Key Words: dimethylformamide (DMF) to produce highly absorbent red chelate with has an absorption maximum Spectrophotometry, Arsenic-Determination, at 505 nm. The absorbance intensity of the metal-chelate reaches a constant value (after heating for 5 2-Hydroxy-1-Napthaldehyde- min at (45±5) 0C within 15 min and remains stable over 24 h. -

System-Friendly Competitive Renewable Energy Procurement in Bangladesh
SYSTEM-FRIENDLY COMPETITIVE RENEWABLE ENERGY PROCUREMENT IN BANGLADESH SCALING UP RENEWABLE ENERGY (SURE) OCTOBER 2020 SYSTEM-FRIENDLY COMPETITIVE RENEWABLE ENERGY PROCUREMENT IN BANGLADESH SCALING UP RENEWABLE ENERGY (SURE) OCTOBER 2020 Prepared for: Energy and Infrastructure Office U.S. Agency for International Development 1300 Pennsylvania Ave NW, Washington DC 20523 Prepared by: Tetra Tech ES, Inc. 1320 North Courthouse Road, Suite 600 Arlington, VA 22201 www.tetratech.com USAID TASK ORDER AID-OAA-I-13-00019AID-OAA-TO-17-00011 DISCLAIMER The views expressed in this publication do not necessarily reflect the views of the United States Agency for International Development or the United States Government. IV SYSTEM-FRIENDLY COMPETITIVE RENEWABLE ENERGY PROCUREMENT IN BANGLADESH Acknowledgements This white paper was prepared by Tetra Tech ES, Inc., and Director, BPDB; Mr. S.M. Zahid Hasan, Deputy Director, Guidehouse (subcontractor). Contributors included Fabian BPDB; Md. Muzibur Rahman, Superintending Engineer, Wigand, Ana Amazo, Bastian Lotz and Tobias Fichter from Dhaka Power Distribution Company (DPDC); Mr. Guidehouse; and Ritesh Kumar Singh, Dr. Alam Hossain Quazi Ashiqur Rahman, Executive Engineer, DPDC; Mr. Mondal and Abu Bakar Siddiq from Tetra Tech. This Abdullah Al Mamun, Head of Project Development Team, white paper is one of the outcomes of USAID’s Scaling Teesta Solar Limited; Mr. Habibur Rahman Faisal, Project Up Renewable Energy (SURE) project in Bangladesh. We Construction Head, Teesta Solar Limited; Mr. Chakma sincerely thank USAID/Bangladesh, which put forth the idea Chandan, Project Engineer, Teesta Solar Limited; Mr. of writing this white paper, with special thanks to Mr. Shayan Ahmed Muntasib Chowdhury, Senior Manager, Engreen Shafi, Senior Energy Advisor, USAID/Bangladesh.