Habitat Heterogeneity Induced by Pyrogenic Organic Matter in Wildfire-Perturbed Soils Mediates Bacterial Community Assembly Processes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Draft Genome Sequence of Acidobacteria Group 1 Acidipila Sp
GENOME SEQUENCES crossm Draft Genome Sequence of Acidobacteria Group 1 Acidipila sp. Strain EB88, Isolated from Forest Soil Luiz A. Domeignoz-Horta,a Kristen M. DeAngelis,a Grace Poldb aDepartment of Microbiology, University of Massachusetts, Amherst, Massachusetts, USA bGraduate Program in Organismic and Evolutionary Biology, University of Massachusetts, Amherst, Massachusetts, USA ABSTRACT Here, we present the genome sequence of a member of the group I Acidobacteria, Acidipila sp. strain EB88, which was isolated from temperate forest soil. Like many other members of its class, its genome contains evidence of the potential to utilize a broad range of sugars. roup I Acidobacteria are abundant members of acidic-soil microbial communities G(1), typically characterized by slow growth, heterotrophy, and the production of copious extracellular polysaccharides (2). Within this group, members of the genus Acidipila are characterized as acidophilic heterotrophs that use sugars as favored growth substrates (3, 4). Acidipila sp. strain EB88 was isolated from the Ͻ0.8-m size fraction of a deciduous forest mineral soil slurry plated onto VL45 plus glucose-yeast extract (GYE) medium (5). It was selected for sequencing based on the paucity of cultivated group I Acidobacteria, its peculiar bright-red waxy colonies on solid medium, and its undetectable growth in liquid medium unless a solid substrate, such as sand, is added. DNA was prepared for sequencing by repeatedly freeze-thawing 9-day-old biomass scraped from pH 5 R2A medium and extracted using the Qiagen genomic DNA protocol. Sequencing at UMass Worcester using the PacBio RS II sequencer resulted in 476,266 filtered reads, with a mean length of 3,208 bp. -

Novel Bacterial Lineages Associated with Boreal Moss Species Hannah
bioRxiv preprint doi: https://doi.org/10.1101/219659; this version posted November 16, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Novel bacterial lineages associated with boreal moss species 2 Hannah Holland-Moritz1,2*, Julia Stuart3, Lily R. Lewis4, Samantha Miller3, Michelle C. Mack3, Stuart 3 F. McDaniel4, Noah Fierer1,2* 4 Affiliations: 5 1Cooperative Institute for Research in Environmental Sciences, University of Colorado at Boulder, 6 Boulder, CO, USA 7 2Department of Ecology and Evolutionary Biology, University of Colorado at Boulder, Boulder, CO, 8 USA 9 3Center for Ecosystem Science and Society, Northern Arizona University, Flagstaff, AZ USA 10 4Department of Biology, University of Florida, Gainesville, FL 32611-8525, USA 11 *Corresponding Author 12 13 Abstract 14 Mosses are critical components of boreal ecosystems where they typically account for a large 15 proportion of net primary productivity and harbor diverse bacterial communities that can be the major 16 source of biologically-fixed nitrogen in these ecosystems. Despite their ecological importance, we have 17 limited understanding of how microbial communities vary across boreal moss species and the extent to 18 which local environmental conditions may influence the composition of these bacterial communities. 19 We used marker gene sequencing to analyze bacterial communities associated with eight boreal moss 20 species collected near Fairbanks, AK USA. We found that host identity was more important than site in 21 determining bacterial community composition and that mosses harbor diverse lineages of potential N2- 22 fixers as well as an abundance of novel taxa assigned to understudied bacterial phyla (including 23 candidate phylum WPS-2). -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

Data of Read Analyses for All 20 Fecal Samples of the Egyptian Mongoose
Supplementary Table S1 – Data of read analyses for all 20 fecal samples of the Egyptian mongoose Number of Good's No-target Chimeric reads ID at ID Total reads Low-quality amplicons Min length Average length Max length Valid reads coverage of amplicons amplicons the species library (%) level 383 2083 33 0 281 1302 1407.0 1442 1769 1722 99.72 466 2373 50 1 212 1310 1409.2 1478 2110 1882 99.53 467 1856 53 3 187 1308 1404.2 1453 1613 1555 99.19 516 2397 36 0 147 1316 1412.2 1476 2214 2161 99.10 460 2657 297 0 246 1302 1416.4 1485 2114 1169 98.77 463 2023 34 0 189 1339 1411.4 1561 1800 1677 99.44 471 2290 41 0 359 1325 1430.1 1490 1890 1833 97.57 502 2565 31 0 227 1315 1411.4 1481 2307 2240 99.31 509 2664 62 0 325 1316 1414.5 1463 2277 2073 99.56 674 2130 34 0 197 1311 1436.3 1463 1899 1095 99.21 396 2246 38 0 106 1332 1407.0 1462 2102 1953 99.05 399 2317 45 1 47 1323 1420.0 1465 2224 2120 98.65 462 2349 47 0 394 1312 1417.5 1478 1908 1794 99.27 501 2246 22 0 253 1328 1442.9 1491 1971 1949 99.04 519 2062 51 0 297 1323 1414.5 1534 1714 1632 99.71 636 2402 35 0 100 1313 1409.7 1478 2267 2206 99.07 388 2454 78 1 78 1326 1406.6 1464 2297 1929 99.26 504 2312 29 0 284 1335 1409.3 1446 1999 1945 99.60 505 2702 45 0 48 1331 1415.2 1475 2609 2497 99.46 508 2380 30 1 210 1329 1436.5 1478 2139 2133 99.02 1 Supplementary Table S2 – PERMANOVA test results of the microbial community of Egyptian mongoose comparison between female and male and between non-adult and adult. -

Tree-Aggregated Predictive Modeling of Microbiome Data
www.nature.com/scientificreports OPEN Tree‑aggregated predictive modeling of microbiome data Jacob Bien1, Xiaohan Yan2, Léo Simpson3,4 & Christian L. Müller4,5,6* Modern high‑throughput sequencing technologies provide low‑cost microbiome survey data across all habitats of life at unprecedented scale. At the most granular level, the primary data consist of sparse counts of amplicon sequence variants or operational taxonomic units that are associated with taxonomic and phylogenetic group information. In this contribution, we leverage the hierarchical structure of amplicon data and propose a data‑driven and scalable tree‑guided aggregation framework to associate microbial subcompositions with response variables of interest. The excess number of zero or low count measurements at the read level forces traditional microbiome data analysis workfows to remove rare sequencing variants or group them by a fxed taxonomic rank, such as genus or phylum, or by phylogenetic similarity. By contrast, our framework, which we call trac (tree‑aggregation of compositional data), learns data‑adaptive taxon aggregation levels for predictive modeling, greatly reducing the need for user‑defned aggregation in preprocessing while simultaneously integrating seamlessly into the compositional data analysis framework. We illustrate the versatility of our framework in the context of large‑scale regression problems in human gut, soil, and marine microbial ecosystems. We posit that the inferred aggregation levels provide highly interpretable taxon groupings that can help microbiome researchers gain insights into the structure and functioning of the underlying ecosystem of interest. Microbial communities populate all major environments on earth and signifcantly contribute to the total plan- etary biomass. Current estimates suggest that a typical human-associated microbiome consists of ∼ 1013 bacteria1 and that marine bacteria and protists contribute to as much as 70% of the total marine biomass2. -

Gut Microbiota Composition Is Correlated to Host Hummingbird Protein Requirements
1 Gut Microbiota Composition is Correlated to Host Hummingbird Protein Requirements ABSTRACT: The gut microbiome shapes and is shaped by a host animal’s physiology. Avian taxa hold physiological characteristics unique from mammals and might inform novel pressures experienced by microbial communities. Further, the symbionts’ relative abundance and their abilities to adapt to available resources are of critical importance to a holobiont’s fitness in rapidly changing climates. Therefore, wild populations of hummingbirds Selasphorus rufus and Calypte anna were studied. The two systems differ in S. rufus’s annual migrations from wintering grounds to their breeding grounds in the Pacific Northwest, whereas C. anna are resident in the latter region. Previous findings have indicated host microbiome composition varied with hummingbird fat score and the month during which fecal sampling occurred. Although fat is an important resource, especially for S. rufus in their migrations, protein requirements are critical because other annual activities, pressuring the organism to access nitrogen. Three of these activities are producing an egg, replacing molted feathers, and carrying parasites. The hypothesis for this study was that birds performing these activities will have microbiota that will make nitrogen available to them. Analysis of OTUs from 16S rDNA V3-V5 amplicon sequencing showed Actinobacteria are more abundant in these hummingbird species than in mammals, replicating our lab’s previous findings. Notably, S. rufus adult females with evidence of recent or current egg production had significantly lower relative levels of Actinobacteria and significantly higher abundances of five of the other ten most abundant bacterial phyla than S. rufus males. The composition of major bacterial phyla in C. -

Microbial Communities of Polymetallic Deposits' Acidic
Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of ANGOR UNIVERSITY Continental Climatic Zone With High Temperature Contrasts Gavrilov, Sergei N.; Korzhenkov, Aleksei A.; Kublanov, Ilya V.; Bargiela, Rafael; Zamana, Leonid V.; Toshchakov, Stepan V.; Golyshin, Peter; Golyshina, Olga Frontiers in Microbiology PRIFYSGOL BANGOR / B Published: 17/07/2019 Publisher's PDF, also known as Version of record Cyswllt i'r cyhoeddiad / Link to publication Dyfyniad o'r fersiwn a gyhoeddwyd / Citation for published version (APA): Gavrilov, S. N., Korzhenkov, A. A., Kublanov, I. V., Bargiela, R., Zamana, L. V., Toshchakov, S. V., Golyshin, P., & Golyshina, O. (2019). Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts. Frontiers in Microbiology. Hawliau Cyffredinol / General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. 01. Oct. 2021 ORIGINAL RESEARCH published: 17 July 2019 doi: 10.3389/fmicb.2019.01573 Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts Sergey N. -
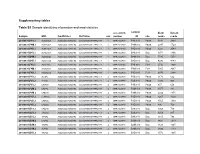
Supplementary Tables Table S1 Sample Identifying Information And
Supplementary tables Table S1 Sample identifying information and read statistics accession sample #raw #clean Sample MID FwdPrimer RvPrimer run number ID site reads reads 20100831.P4S.1 ACACGAGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4S.910 Palsa 3831 2893 20100831.P4M.1 ACATAGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4M.910 Palsa 2047 1729 20100831.P4D.1 ACGATACA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4D.910 Palsa 3221 2543 20100831.B4S.1 ACTGCTCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4S.910 Bog 1971 1366 20100831.B4M.1 AGCAGCGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4M.910 Bog 5530 5251 20100831.B4D.1 AGCGTGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4D.910 Bog 4296 4143 20100831.F4S.1 AGTCTAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4S.910 Fen* 2716 1890 20100831.F4M.1 ATGACTGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4M.910 Fen* 5005 3987 20100831.F4D.1 ATGCACGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4D.910 Fen* 2870 2384 20100901.P1S.2 ACTGAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P1S.910 Palsa 1170 662 20100901.P2S.2 ATGTGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2S.910 Palsa 1320 923 20100901.P3S.2 CACAGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3S.910 Palsa 857 656 20100901.P2M.2 CACTAC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2M.910 Palsa 1077 881 20100901.P3M.2 CAGCAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3M.910 Palsa 2032 1474 20100901.P1D.2 CTAGAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT -

Metagenomic SMRT Sequencing-Based Exploration Of
J. Microbiol. Biotechnol. (2017), 27(9), 1670–1680 https://doi.org/10.4014/jmb.1705.05008 Research Article Review jmb Metagenomic SMRT Sequencing-Based Exploration of Novel Lignocellulose-Degrading Capability in Wood Detritus from Torreya nucifera in Bija Forest on Jeju Island Han Na Oh1, Tae Kwon Lee2, Jae Wan Park1, Jee Hyun No2, Dockyu Kim3, and Woo Jun Sul1* 1Department of Systems Biotechnology, Chung-Ang University, Anseong 17546, Republic of Korea 2Department of Environmental Engineering, Yonsei University, Wonju 26493, Republic of Korea 3Division of Life Sciences, Korea Polar Research Institute, Incheon 21990, Republic of Korea Received: May 4, 2017 Revised: June 12, 2017 Lignocellulose, composed mostly of cellulose, hemicellulose, and lignin generated through Accepted: June 19, 2017 secondary growth of woody plant, is considered as promising resources for biofuel. In order to First published online use lignocellulose as a biofuel, biodegradation besides high-cost chemical treatments were June 21, 2017 applied, but knowledge on the decomposition of lignocellulose occurring in a natural *Corresponding author environment is insufficient. We analyzed the 16S rRNA gene and metagenome to understand Phone: +82-31-670-4707; how the lignocellulose is decomposed naturally in decayed Torreya nucifera (L) of Bija forest Fax: +82-31-670-3108; E-mail: [email protected] (Bijarim) in Gotjawal, an ecologically distinct environment. A total of 464,360 reads were obtained from 16S rRNA gene sequencing, representing diverse phyla; Proteobacteria (51%), Bacteroidetes (11%) and Actinobacteria (10%). The metagenome analysis using single molecules real-time sequencing revealed that the assembled contigs determined originated from Proteobacteria (58%) and Actinobacteria (10.3%). -

Pyrosequencing Investigation Into the Bacterial Community in Permafrost Soils Along the China-Russia Crude Oil Pipeline (CRCOP)
Pyrosequencing Investigation into the Bacterial Community in Permafrost Soils along the China-Russia Crude Oil Pipeline (CRCOP) Sizhong Yang1*, Xi Wen2, Huijun Jin1, Qingbai Wu1 1 State Key Laboratory of Frozen Soil Engineering (SKLFSE), Cold and Arid Regions Environmental and Engineering Research Institute (CAREERI), Chinese Academy of Sciences, Lanzhou, China, 2 College of Electrical Engineering, Northwest University for Nationalities, Lanzhou, China Abstract The China-Russia Crude Oil Pipeline (CRCOP) goes through 441 km permafrost soils in northeastern China. The bioremediation in case of oil spills is a major concern. So far, little is known about the indigenous bacteria inhabiting in the permafrost soils along the pipeline. A pilot 454 pyrosequencing analysis on the communities from four selected sites which possess high environment risk along the CRCOP is herein presented. The results reveal an immense bacterial diversity than previously anticipated. A total of 14448 OTUs with 84834 reads are identified, which could be assigned into 39 different phyla, and 223 families or 386 genera. Only five phyla sustain a mean OTU abundance more than 5% in all the samples, but they altogether account for 85.08% of total reads. Proteobacteria accounts for 41.65% of the total OTUs or 45% of the reads across all samples, and its proportion generally increases with soil depth, but OTUs numerically decline. Among Proteobacteria, the abundance of Beta-, Alpha-, Delta- and Gamma- subdivisions average to 38.7% (2331 OTUs), 37.5% (2257 OTUs), 10.35% (616 OTUs), and 6.21% (374 OTUs), respectively. Acidobacteria (esp. Acidobacteriaceae), Actinobacteria (esp. Intrasporangiaceae), Bacteroidetes (esp. Sphingobacteria and Flavobacteria) and Chloroflexi (esp. -

Studying the Microbiome of Cyanobacterial Biocrusts from Drylands and Its Functional Infuence on Biogeochemical Cycles
Studying the Microbiome of Cyanobacterial Biocrusts From Drylands and Its Functional Inuence on Biogeochemical Cycles Isabel Miralles ( [email protected] ) University of Almería Raúl Ortega University of Almería Maria Carmen Montero-Calasanz Newcastle University Research Article Keywords: desert biome, soil bacterial taxa, shotgun, bacterial functions, MG-RAST, MicrobiomeAnalyst Posted Date: February 24th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-252045/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/51 Abstract Background: Drylands are areas under continuous degradation and desertication largely covered by cyanobacterial biocrusts. Several studies have already shown that soil microorganisms play a fundamental role in the correct soil functioning. Nevertheless, little is known about the relationship taxonomy-function in arid soils and, in particular, in cyanobacterial biocrusts. An in-depth study of the taxonomic composition and the functions carried out by soil microorganisms in biogeochemical cycles was here carried out by using a shotgun metagenomic approach. Results: Metagenomic analysis carried out in this study showed a high taxonomic and functional similarity in both incipient and mature cyanobacterial biocrusts types with a dominance of Proteobacteria, Actinobacteria and Cyanobacteria. The predominant functional categories related to soil biogeochemical cycles were “carbon metabolism” followed by “phosphorus, nitrogen, sulfur, potassium and iron metabolism”. Reads involved in the metabolism of carbohydrates and respiration were the most abundant functional classes. In the N cycle dominated “ammonia assimilation” and “Nitrate and nitrite ammonication”. The major taxonomic groups also seemed to drive phosphorus and potassium cycling by the production of organic acids and the presence of extracellular enzymes and specialised transporters. -

Molecular Taxonomy and Community Dynamics of Actinobacteria In
Revista de Biología Marina y Oceanografía Vol. 51, Nº1: 89-100, abril 2016 DOI 10.4067/S0718-19572016000100009 ARTICLE Molecular taxonomy and community dynamics of Actinobacteria in marine sediments off central Chile Taxonomía molecular y dinámica comunitaria de Actinobacteria en sedimentos marinos de Chile central Selim S. Musleh1, Daniel Gomez-Uchida2, Carola Espinoza1, Nathaly Ruiz-Tagle3, Alexis Fonseca1 and Víctor A. Gallardo1 1Department of Oceanography, Universidad de Concepción, P.O. Box 160-C, Concepción, Chile. [email protected] 2Department of Zoology, Universidad de Concepción, P.O. Box 160- C, Concepción, Chile 3Centro de Biotecnología, Universidad de Concepción, P.O. Box 160- C, Concepción, Chile Resumen.- Se usó pirosecuenciación de la región V6 del gen 16S del ARNr para caracterizar la diversidad y la dinámica espacio- temporal de unidades taxonómicas operacionales (OTUs) del filo Actinobacteria, los que fueron aislados desde sedimentos provenientes del Sulfureto de Humboldt frente a Chile central. Este substrato es rico en compuestos azufrados y material orgánico lo que mantiene una vasta comunidad microbiana que experimenta cambios estacionales en respuesta a regímenes oceanográficos contrastantes. Se identificaron 498 OTUs distribuidas en 7 órdenes, 47 familias, 122 géneros, (5 de los cuales son ampliamente reconocidos por sus aplicaciones biotecnológicas), y 56 especies. El análisis temporal reveló que algunos OTUs presentan diferencias significativas en abundancia, índices de diversidad y riqueza, las que generaron una agrupación de las muestras asociada a la fecha de muestreo (estación del año) y no a la profundidad del sitio de muestreo. Debido a que las Actinobacteria son mayormente aeróbicas, las altas concentraciones de oxígeno disuelto que ocurren en la zona en el otoño-invierno austral, representan condiciones ambientales beneficiosas para este filo, no así las de primavera-verano austral cuando prevalece la hipoxia.