Amino Acids and Peptides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subject Index
Cambridge University Press 0521462924 - Amino Acids and Peptides G. C. Barrett and D. T. Elmore Index More information Subject index acetamidomalonate synthesis 123–4 in fossil dating 15 S-adenosyl--methionine 11, 12, 174, 181 in geological samples 15 alanine, N-acetyl 9 in Nature 1 structure 4 IR spectrometry 36 alkaloids, biosynthesis from amino acids 16–17 isolation from proteins 121 alloisoleucine 6 mass spectra 61 allosteric change 178 metabolism, products of 187 allothreonine 6 metal-binding properties 34 Alzheimer’s disease 14 NMR 41 amidation at peptide C-terminus 57, 94, 181 physicochemical properties 32 amides, cis–trans isomerism 20 protein 3 N-acylation 72 PTC derivatives 87 N-alkylation 72 quaternary ammonium salts 50 hydrolysis 57 reactions of amino group 49, 51 O-trimethylsilylation 72 reactions of carboxy group 49, 53 reduction to -aminoalkanols 72 racemisation 56 amidocarbonylation in amino acid synthesis 125 routine spectrometry 35 amino acids, abbreviated names 7 Schöllkopf synthesis 127–8 acid–base properties 32 Schiff base formation 49 antimetabolites 200 sequence determination 97 et seq. as food additives 14 following selective chemical degradation 107 as neurotransmitters 17 following selective enzymic degradation 109 asymmetric synthesis 127 general strategy for 92 - 17–18 identification of C-terminus 106–7 biosynthesis 121 identification of N-terminus 94, 97 biosynthesis from, of creatinine 183 by solid-phase methodology 100 of nitric oxide 186 by stepwise chemical degradation 97 of porphyrins 185 by use of dipeptidyl -

Designing Peptidomimetics
CORE Metadata, citation and similar papers at core.ac.uk Provided by UPCommons. Portal del coneixement obert de la UPC DESIGNING PEPTIDOMIMETICS Juan J. Perez Dept. of Chemical Engineering ETS d’Enginyeria Industrial Av. Diagonal, 647 08028 Barcelona, Spain 1 Abstract The concept of a peptidomimetic was coined about forty years ago. Since then, an enormous effort and interest has been devoted to mimic the properties of peptides with small molecules or pseudopeptides. The present report aims to review different approaches described in the past to succeed in this goal. Basically, there are two different approaches to design peptidomimetics: a medicinal chemistry approach, where parts of the peptide are successively replaced by non-peptide moieties until getting a non-peptide molecule and a biophysical approach, where a hypothesis of the bioactive form of the peptide is sketched and peptidomimetics are designed based on hanging the appropriate chemical moieties on diverse scaffolds. Although both approaches have been used in the past, the former has been more widely used to design peptidomimetics of secretory peptides, whereas the latter is nowadays getting momentum with the recent interest in designing protein-protein interaction inhibitors. The present report summarizes the relevance of the information gathered from structure-activity studies, together with a short review on the strategies used to design new peptide analogs and surrogates. In a following section there is a short discussion on the characterization of the bioactive conformation of a peptide, to continue describing the process of designing conformationally constrained analogs producing first and second generation peptidomimetics. Finally, there is a section devoted to review the use of organic scaffolds to design peptidomimetics based on the information available on the bioactive conformation of the peptide. -

Polyketide Synthase Gene Coupled to the Peptide Synthetase Module Involved in the Biosynthesis of the Cyclic Heptapeptide Microcystinl
J. Biochem. 127, 779-789(2000) Polyketide Synthase Gene Coupled to the Peptide Synthetase Module Involved in the Biosynthesis of the Cyclic Heptapeptide Microcystinl Tomoyasu Nishizawa, Akiko Ueda, Munehiko Asayama,* Kiyonaga Fujii,•õ Ken-ichi Harada,i Kozo Ochi,•ö and Makoto Shirai*,•˜,2 * Division of Biotechnology, School ofAgriculture, Ibaraki University Ami , Ibaraki 300-0393; •õFaculty of Pharmacy, M eija University, Tempaku , Nagoya 468-8503, •öNational Food Research Institute, Tsukuba, Ibaraki 305-8642; and •˜ Gene Research Center, Ibaraki University, Ann, Ibaraki 300-0393 Received January 17, 2000; accepted February 11, 2000 The peptide synthetase gene operon, which consists of encyA, mcyB, and mcyC, for the activation and incorporation of the five amino acid constituents of microcystin has been identified [T. Nishizawa et al. (1999) J. Biochem. 126, 520-529] . By sequencing an addi tional 34 kb of DNA from microcystin-producing Microcystis aeruginosa K-139 , we identifi ed the residual microcystin synthetase gene operon, which consists of mcyD, mcyE, meyF, and mcyG, in the opposite orientation to the mcyABC operon. McyD consisted of two polyketide synthase modules, and McyE contained a polyketide synthase module at the N-terminus and a peptide synthetase module at the C-terminus. McyF was found to exhibit similarity to amino acid racemase. McyG consisted of a peptide synthetase mod ule at the N-terminus and a polyketide synthase at the C-terminus. The microcystin syn thetase gene cluster was conserved in another microcystin-producing strain, Microcystis sp. S-70, which produces Microcystin-LR, -RR, and -YR. Insertional mutagenesis of mcyA, mcyD, or meyE in Microcystis sp. -

Interrupted Adenylation Domains: Unique Bifunctional Enzymes Involved in Nonribosomal Peptide Biosynthesis
Natural Product Reports Interrupted adenylation domains: unique bifunctional enzymes involved in nonribosomal peptide biosynthesis Journal: Natural Product Reports Manuscript ID: NP-REV-09-2014-000120.R1 Article Type: Highlight Date Submitted by the Author: 12-Jan-2015 Complete List of Authors: Labby, Kristin; Beloit College, Chemistry Watsula, Stoyan; University of Michigan, Medicinal Chemistry Garneau-Tsodikova, Sylvie; University of Kentucky, Pharmaceutical Sciences Page 1 of 11NPR Natural Product Reports Dynamic Article Links ► Cite this: DOI: 10.1039/c0xx00000x www.rsc.org/xxxxxx HIGHLIGHT Interrupted adenylation domains: unique bifunctional enzymes involved in nonribosomal peptide biosynthesis Kristin J. Labby, a Stoyan G. Watsula,b and Sylvie Garneau-Tsodikova* c Received (in XXX, XXX) Xth XXXXXXXXX 20XX, Accepted Xth XXXXXXXXX 20XX 5 DOI: 10.1039/b000000x Covering up to 2014 Nonribosomal peptides (NRPs) account for a large portion of drugs and drug leads currently available in the pharmaceutical industry. They are one of two main families of natural products biosynthesized on megaenzyme assembly-lines composed of multiple modules that are, in general, each comprised of three 10 core domains and on occasion of accompanying auxiliary domains. The core adenylation (A) domains are known to delineate the identity of the specific chemical components to be incorporated into the growing NRPs. Previously believed to be inactive, A domains interrupted by auxiliary enzymes have recently been proven to be active and capable of performing two distinct chemical reactions. This highlight summarizes current knowledge on A domains and presents the various interrupted A domains found in a number of 15 nonribosomal peptide synthetase (NRPS) assembly-lines, their predicted or proven dual functions, and their potential for manipulation and engineering for chemoenzymatic synthesis of new pharmaceutical agents with increased potency. -

COMPILATION of AMINO ACIDS, DRUGS, METABOLITES and OTHER COMPOUNDS in MASSTRAK AMINO ACID ANALYSIS SOLUTION Paula Hong, Kendon S
COMPILATION OF AMINO ACIDS, DRUGS, METABOLITES AND OTHER COMPOUNDS IN MASSTRAK AMINO ACID ANALYSIS SOLUTION Paula Hong, Kendon S. Graham, Alexandre Paccou, T homas E. Wheat and Diane M. Diehl INTRODUCTION LC conditions Physiological amino acid analysis is commonly performed to LC System: Waters ACQUITY UPLC® System with TUV monitor and study a wide variety of metabolic processes. A wide Column: MassTrak AAA Column 2.1 x 150 mm, 1.7 µm variety of drugs, foods, and metabolic intermediates that may Column Temp: 43 ˚C be present in biological fluids can appear as peaks in amino Flow Rate: 400 µL/min. acid analysis, therefore, it is important to be able to identify Mobile Phase A: MassTrak AAA Eluent A Concentrate, unknown compounds.1,2,3 The reproducibility and robustness of diluted 1:10 the MassTrak Amino Acid Analysis Solution make this method well Mobile Phase B: MassTrak AAA Eluent B suited to such a study as well.4 Weak Needle Wash: 5/95 Acetonitrile/Water Strong Needle Wash: 95/5 Acetonitrile/Water Gradient: MassTrak AAA Standard Gradient (as provided in kit) Detection: UV @ 260 nm Injection Volume: 1 µL EXPERIMENTAL Injection Mode: Partial Loop with Needle Overfill (PLNO) Compound sample preparation A library of compounds was assembled. Each compound was derivatized individually and spiked into the MassTrak™ AAA Solution Standard prior to chromatographic analysis. The elution RESULTS AND DISCUSSION position of each tested compound could be related to known amino acids. A wide variety of antibiotics, pharmaceutical compounds and metabolite by-products are found in biological fluids. The reten- 1. -
Supplementary File 1 (PDF, 650 Kib)
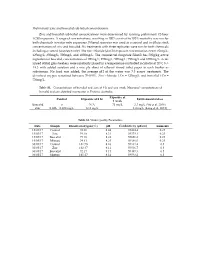
Preliminary Zinc and boscalid sub lethal concentrations Zinc and boscalid sub lethal concentrations were determined by running preliminary 72‐hour LC50 exposures. A range of concentrations, resulting in 100% survival to 100% mortality was run for both chemicals in water only exposures. Filtered seawater was used as a control and to dilute stock concentrations of zinc and boscalid. Six treatments with three replicates were run for both chemicals including a control (seawater only). The zinc chloride (ZnCl2) exposure concentrations were: 62mg/L; 125mg/L; 250mg/L; 500mg/L and 1000mg/L. The commercial fungicide Filan® has 500g/kg active ingredient of boscalid, concentrations of 100mg/L; 250mg/L; 500mg/L; 750mg/L and 1000mg/L. Acid‐ rinsed 600ml glass beakers were randomly placed in a temperature‐controlled incubator at 20oC (+/‐ 1oC) with added aeration and a one ply sheet of ethanol rinsed toilet paper in each beaker as substratum. No food was added; the average pH of the water was 7.5 across treatments. The dissolved oxygen remained between 70‐100%. Zinc chloride LC50 = 125mg/L and boscalid LC50 = 750mg/L. Table S1. –Concentrations of boscalid and zinc at 0 h and one week. Measured concentrations of boscalid and zinc detected in estuaries in Victoria, Australia. Exposure at Control Exposure at 0 hr Environmental dose 1 week Boscalid 0 N/A 75 mg/L 3.3 mg/L (Vu et al. 2016) Zinc 0.026 – 0.034 mg/L 12.5 mg/L 3.4 mg/L (Long et al. 2015) Table S2. Water Quality Parameters. Date Sample Dissolved oxygen (%) pH Conductivity (µS/cm) Ammonia 15/05/17 Control 98.36 8.44 59464.4 0.25 15/05/17 Zinc 99.10 8.31 59379.1 0.25 15/05/17 Boscalid 99.18 8.28 59656.8 0.25 15/05/17 Mixture 94.81 8.25 59386.0 0.25 30/05/17 Control 101.90 8.16 59332.4 0.5 30/05/17 Zinc 102.47 8.11 59536.7 0.5 30/05/17 Boscalid 92.17 8.15 59489.3 0.5 30/05/17 Mixture 101.57 8.14 59593.2 0.5 Supplementary Material . -

N,N-Diprotected Dehydroamino Acid Derivatives: Versatile Substrates for the Synthesis of Novel Amino Acids
N,N-DIPROTECTED DEHYDROAMINO ACID DERIVATIVES: VERSATILE SUBSTRATES FOR THE SYNTHESIS OF NOVEL AMINO ACIDS Paula M. T. Ferreira and Luís S. Monteiro Department of Chemistry, University of Minho, Gualtar, 4710-057 Braga, Portugal (e-mail: [email protected]) Abstract. Non-proteinogenic amino acids are an important class of organic compounds that can have intrinsic biological activity or can be found in peptides with antiviral, antitumor, anti-inflammatory or immunosuppressive activities. This type of compounds is also important in drug development, in the elucidation of biochemical pathways and in conformational studies. Therefore, research towards efficient methods that allow the synthesis of these compounds constitutes an important area of peptide chemistry. In our laboratories we have developed a new and high yielding method for the synthesis of N,N-diprotected dehydroamino acid derivatives using tert-butyl pyrocarbonate and 4-dimethylaminopyridine. These compounds were used as substrates in several types of reactions, allowing the synthesis of a variety of new amino acid derivatives. Some of these new compounds are heterocyclic systems or contain heterocyclic moieties such as pyrazole, indole, or imidazole. Thus, several nitrogen heterocycles were reacted with N,N-diprotected dehydroalanine to give new β-substituted alanines and dehydroalanines. Furanic amino acids were obtained treating the methyl ester of N-(4-toluenesulfonyl), N-(tert-butoxycarbonyl) dehydroalanine with carbon nucleophiles of the β-dicarbonyl type having at least one methyl group attached to one of the carbonyl groups. Treatment of these furanic amino acids with trifluoracetic acid afforded pyrrole derivatives in good to high yields. A N,N-diprotected 1,4-dihydropyrazine was obtained reacting the methyl ester of N-(4-toluenesulfonyl), N-(tert-butoxycarbonyl)dehydroalanine with 4-dimethylaminopyridine and an excess of potassium carbonate. -

Phenotype Microarrays™
Phenotype MicroArrays™ PM1 MicroPlate™ Carbon Sources A1 A2 A3 A4 A5 A6 A7 A8 A9 A10 A11 A12 Negative Control L-Arabinose N-Acetyl -D- D-Saccharic Acid Succinic Acid D-Galactose L-Aspartic Acid L-Proline D-Alanine D-Trehalose D-Mannose Dulcitol Glucosamine B1 B2 B3 B4 B5 B6 B7 B8 B9 B10 B11 B12 D-Serine D-Sorbitol Glycerol L-Fucose D-Glucuronic D-Gluconic Acid D,L -α-Glycerol- D-Xylose L-Lactic Acid Formic Acid D-Mannitol L-Glutamic Acid Acid Phosphate C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 C11 C12 D-Glucose-6- D-Galactonic D,L-Malic Acid D-Ribose Tween 20 L-Rhamnose D-Fructose Acetic Acid -D-Glucose Maltose D-Melibiose Thymidine α Phosphate Acid- -Lactone γ D-1 D2 D3 D4 D5 D6 D7 D8 D9 D10 D11 D12 L-Asparagine D-Aspartic Acid D-Glucosaminic 1,2-Propanediol Tween 40 -Keto-Glutaric -Keto-Butyric -Methyl-D- -D-Lactose Lactulose Sucrose Uridine α α α α Acid Acid Acid Galactoside E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 E11 E12 L-Glutamine m-Tartaric Acid D-Glucose-1- D-Fructose-6- Tween 80 -Hydroxy -Hydroxy -Methyl-D- Adonitol Maltotriose 2-Deoxy Adenosine α α ß Phosphate Phosphate Glutaric Acid- Butyric Acid Glucoside Adenosine γ- Lactone F1 F2 F3 F4 F5 F6 F7 F8 F9 F10 F11 F12 Glycyl -L-Aspartic Citric Acid myo-Inositol D-Threonine Fumaric Acid Bromo Succinic Propionic Acid Mucic Acid Glycolic Acid Glyoxylic Acid D-Cellobiose Inosine Acid Acid G1 G2 G3 G4 G5 G6 G7 G8 G9 G10 G11 G12 Glycyl-L- Tricarballylic L-Serine L-Threonine L-Alanine L-Alanyl-Glycine Acetoacetic Acid N-Acetyl- -D- Mono Methyl Methyl Pyruvate D-Malic Acid L-Malic Acid ß Glutamic Acid Acid -

Structural Characterization of Microcystins by LC/MS/MS Under
J. Antibiot. 59(11): 710–719, 2006 THE JOURNAL OF ORIGINAL ARTICLE ANTIBIOTICS Structural Characterization of Microcystins by LC/MS/MS under Ion Trap Conditions Tsuyoshi Mayumi, Hajime Kato, Susumu Imanishi, Yoshito Kawasaki, Masateru Hasegawa, Ken-ichi Harada This article is dedicated in memory of Professor Kenneth L. Rinehart at the University of Illinois Received: May 16, 2006 / Accepted: November 2, 2006 © Japan Antibiotics Research Association Abstract LC/MS/MS under ion trap conditions was used cyclic peptide antibiotics such as bacitracin [1], colistin [2], to analyze microcystins produced by cyanobacteria. vancomycin [3] and micafungin [4], are widely used for Tandem mass spectrometry using MS2 was quite effective medical treatments. Moreover, cyclosporin [5] is an since ions arising from cleavage at a peptide bond provide immunosuppressive agent, which has been used for organ useful sequence information. The fragmentation was transplant rejection or bone marrow [6]. confirmed by a shifting technique using structurally-related For the structure determination of cyclic peptides, the microcystins and the resulting fragmentation pattern was following information is required: structures, absolute different from those determined by triple stage MS/MS and configurations and sequence of constituent amino acids. four sector MS/MS. Analysis of a mixture of microcystins Although partial acid hydrolysis had been used for the in a bloom sample was successfully performed and two structure determination of these cyclic peptides, it has been new microcystins were identified by LC/MS/MS under ion currently performed by instrumental methods such as 2D- trap conditions. Thus, LC/MS/MS under ion trap NMR (two dimensional nuclear magnetic resonance) and conditions is effective for the structural characterization of MS/MS (tandem mass spectrometry) techniques. -

These Lanthionine FINALE
FACULTÉ DES SCIENCES – DÉPARTEMENT DE CHIMIE CENTRE DE RECHERCHES DU CYCLOTRON SYNTHESIS AND BIOLOGICAL STUDIES OF LANTHIONINE DERIVATIVES Promoteur: Professeur André LUXEN Dissertation présentée par Thibaut DENOËL pour l’obtention du grade de Docteur en Sciences Année académique 2013 - 2014 Membres du Jury : Promoteur : Professeur André Luxen (Université de Liège) Président : Professeur Albert Demonceau (Université de Liège) Autres membres : Docteur Didier Blanot (Université Paris-Sud) Docteur David Thonon (Uteron Pharma) Professeur Bernard Joris (Université de Liège) Docteur Christian Lemaire (Université de Liège) Remerciements Les travaux présentés dans cette thèse ont été réalisés sous la direction du Professeur André Luxen dans les laboratoires du Centre de Recherches du Cyclotron de l’Université de Liège. Je voudrais tout d’abord remercier le Professeur André Luxen pour son accueil au sein du Cyclotron, sa grande patience et son exigence. Il a aussi inspiré nombre de manipulations et m’a guidé dans mes recherches. En outre, il a mis à ma disposition tout le matériel et la quantité de produits chimiques nécessaires à la réalisation de ce travail. Je tiens également à remercier Messieurs les membres du Jury pour m’avoir fait l’honneur d’accepter d’examiner ce travail, en l’occurrence le Docteur Didier Blanot (Université Paris-Sud), le Docteur David Thonon (Uteron Pharma), le Professeur Bernard Joris, le Docteur Christian Lemaire et le Professeur Albert Demonceau dans le rôle de Président. Je remercie aussi les Docteurs Didier Blanot et Mireille Hervé pour la réalisation des essais in vitro avec l’enzyme Mpl et l’aide dans les publications. Je remercie grandement le Docteur Astrid Zervosen pour la réalisation des manipulations biochimiques, ses conseils, sa disponibilité et la correction lors de la rédaction. -

Microcystin-LR Toxicodynamics, Induced Pathology, and Immunohistochemical Localization in Livers of Blue-Green Algae Exposed Rainbow Trout (Oncorhynchus Mykiss)
Microcystin-LR Toxicodynamics, Induced Pathology, and Immunohistochemical Localization in Livers of Blue-Green Algae Exposed Rainbow Trout (Oncorhynchus mykiss) W. J. Fischer,* B. C. Hitzfeld,* F. Tencalla,† J. E. Eriksson,‡ A. Mikhailov,‡ and D. R. Dietrich*,1 *Environmental Toxicology, University of Konstanz, Konstanz, Germany; †Institute of Toxicology, Schwerzenbach, Switzerland; and ‡Turku Centre for Biotechnology, Turku, Finland Received June 29, 1999; accepted September 27, 1999 Microcystins (MC) constitute a family of toxins that are With this retrospective study, we investigated the temporal produced by several cyanobacterial taxa. These cyclic hep- pattern of toxin exposure and pathology, as well as the topical tapeptide molecules contain both L- and D-amino acids and an relationship between hepatotoxic injury and localization of micro- unusual hydrophobic C D-amino acid commonly termed cystin-LR, a potent hepatotoxin, tumor promoter, and inhibitor of 20 protein phosphatases-1 and -2A (PP), in livers of MC-gavaged ADDA (3-amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca- rainbow trout (Oncorhynchus mykiss) yearlings, using an immu- 4,6-dienoic acid). In most of the more-than-60 presently known nohistochemical detection method and MC-specific antibodies. toxin congeners, the 5 D-amino acid components are main- H&E stains of liver sections were used to determine pathological tained while the two L-amino acids are variable (Botes et al., changes. Nuclear morphology of hepatocytes and ISEL analysis 1985). Microcystin-LR, containing L-Leu and L-Arg, is one of were employed as endpoints to detect the advent of apoptotic cell the most commonly occurring (Watanabe et al., 1996) and at death in hepatocytes. -

Amino Acid - Wikipedia, the Free Encyclopedia
1/29/2016 Amino acid - Wikipedia, the free encyclopedia Amino acid From Wikipedia, the free encyclopedia This article is about the class of chemicals. For the structures and properties of the standard proteinogenic amino acids, see Proteinogenic amino acid. Amino acidsarebiologically important organic compoundscontaining amine (-NH2) and carboxylic acid(- COOH) functional groups, usually along with a side-chain specific to each aminoacid.[1][2][3] The key elements of an amino acid are carbon, hydrogen, oxygen, andnitrogen, though other elements are found in the side-chains of certain amino acids. About 500 amino acids are known and can be classified in many ways.[4] They can be classified according to the core structural functional groups' locations as alpha- (α-), beta- (β-), gamma- (γ-) or delta- (δ-) amino acids; other categories relate to polarity, pHlevel, and side-chain group type (aliphatic,acyclic, aromatic, containing hydroxyl orsulfur, etc.). In the form of proteins, amino acids comprise the second-largest component (water is the largest) of humanmuscles, cells and other tissues.[5] Outside proteins, The structure of an alpha amino amino acids perform critical roles in processes such as neurotransmittertransport and biosynthesis. acid in its un-ionized form In biochemistry, amino acids having both the amine and the carboxylic acid groups attached to the first (alpha-) carbon atom have particular importance. They are known as 2-, alpha-, or α-amino [6] acids (genericformula H2NCHRCOOH in most cases, where R is an organic substituent