Gene Co-Expression Networks Contributing to the Expression Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Chromosomal Aberrations in Head and Neck Squamous Cell Carcinomas in Norwegian and Sudanese Populations by Array Comparative Genomic Hybridization
825-843 12/9/08 15:31 Page 825 ONCOLOGY REPORTS 20: 825-843, 2008 825 Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization ERIC ROMAN1,2, LEONARDO A. MEZA-ZEPEDA3, STINE H. KRESSE3, OLA MYKLEBOST3,4, ENDRE N. VASSTRAND2 and SALAH O. IBRAHIM1,2 1Department of Biomedicine, Faculty of Medicine and Dentistry, University of Bergen, Jonas Lies vei 91; 2Department of Oral Sciences - Periodontology, Faculty of Medicine and Dentistry, University of Bergen, Årstadveien 17, 5009 Bergen; 3Department of Tumor Biology, Institute for Cancer Research, Rikshospitalet-Radiumhospitalet Medical Center, Montebello, 0310 Oslo; 4Department of Molecular Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway Received January 30, 2008; Accepted April 29, 2008 DOI: 10.3892/or_00000080 Abstract. We used microarray-based comparative genomic logical parameters showed little correlation, suggesting an hybridization to explore genome-wide profiles of chromosomal occurrence of gains/losses regardless of ethnic differences and aberrations in 26 samples of head and neck cancers compared clinicopathological status between the patients from the two to their pair-wise normal controls. The samples were obtained countries. Our findings indicate the existence of common from Sudanese (n=11) and Norwegian (n=15) patients. The gene-specific amplifications/deletions in these tumors, findings were correlated with clinicopathological variables. regardless of the source of the samples or attributed We identified the amplification of 41 common chromosomal carcinogenic risk factors. regions (harboring 149 candidate genes) and the deletion of 22 (28 candidate genes). Predominant chromosomal alterations Introduction that were observed included high-level amplification at 1q21 (harboring the S100A gene family) and 11q22 (including Head and neck squamous cell carcinoma (HNSCC), including several MMP family members). -
Download Validation Data
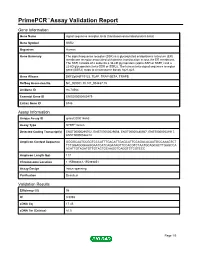
PrimePCR™Assay Validation Report Gene Information Gene Name signal sequence receptor, beta (translocon-associated protein beta) Gene Symbol SSR2 Organism Human Gene Summary The signal sequence receptor (SSR) is a glycosylated endoplasmic reticulum (ER) membrane receptor associated with protein translocation across the ER membrane. The SSR consists of 2 subunits a 34-kD glycoprotein (alpha-SSR or SSR1) and a 22-kD glycoprotein (beta-SSR or SSR2). The human beta-signal sequence receptor gene (SSR2) maps to chromosome bands 1q21-q23. Gene Aliases DKFZp686F19123, TLAP, TRAP-BETA, TRAPB RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.74564 Ensembl Gene ID ENSG00000163479 Entrez Gene ID 6746 Assay Information Unique Assay ID qHsaCID0014663 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000295702, ENST00000529008, ENST00000480567, ENST00000531917, ENST00000526212 Amplicon Context Sequence GGGGCAATCCGGTCCCATTTGACATTGAGCATTCCAGACACAATGCCAAAGTCT TCTGGAGGGAAGGAATCATCAGATAGTTCCACGTCTAATGCAGCACTTGAGCCA ACATTGTAGATGTTGTACTGCAAGGTCAGGTCTCGTCCC Amplicon Length (bp) 117 Chromosome Location 1:155988061-155989851 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9998 cDNA Cq 17.45 cDNA Tm (Celsius) 81.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SSR2, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

Reporterseq Reveals Genome-Wide Dynamic Modulators of the Heat
RESEARCH ARTICLE ReporterSeq reveals genome-wide dynamic modulators of the heat shock response across diverse stressors Brian D Alford1†, Eduardo Tassoni-Tsuchida1,2†, Danish Khan1, Jeremy J Work1, Gregory Valiant3, Onn Brandman1* 1Department of Biochemistry, Stanford University, Stanford, United States; 2Department of Biology, Stanford University, Stanford, United States; 3Department of Computer Science, Stanford University, Stanford, United States Abstract Understanding cellular stress response pathways is challenging because of the complexity of regulatory mechanisms and response dynamics, which can vary with both time and the type of stress. We developed a reverse genetic method called ReporterSeq to comprehensively identify genes regulating a stress-induced transcription factor under multiple conditions in a time- resolved manner. ReporterSeq links RNA-encoded barcode levels to pathway-specific output under genetic perturbations, allowing pooled pathway activity measurements via DNA sequencing alone and without cell enrichment or single-cell isolation. We used ReporterSeq to identify regulators of the heat shock response (HSR), a conserved, poorly understood transcriptional program that protects cells from proteotoxicity and is misregulated in disease. Genome-wide HSR regulation in budding yeast was assessed across 15 stress conditions, uncovering novel stress-specific, time- specific, and constitutive regulators. ReporterSeq can assess the genetic regulators of any transcriptional pathway with the scale of pooled genetic screens and the precision of pathway- specific readouts. *For correspondence: [email protected] †These authors contributed equally to this work Introduction Competing interests: The The heat shock response (HSR) is a conserved stress response that shields cells from cytoplasmic authors declare that no proteotoxicity by increasing the expression of protective proteins (Lindquist, 1986; Mori- competing interests exist. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Ncomms4301.Pdf
ARTICLE Received 8 Jul 2013 | Accepted 23 Jan 2014 | Published 13 Feb 2014 DOI: 10.1038/ncomms4301 Genome-wide RNAi ionomics screen reveals new genes and regulation of human trace element metabolism Mikalai Malinouski1,2, Nesrin M. Hasan3, Yan Zhang1,4, Javier Seravalli2, Jie Lin4,5, Andrei Avanesov1, Svetlana Lutsenko3 & Vadim N. Gladyshev1 Trace elements are essential for human metabolism and dysregulation of their homoeostasis is associated with numerous disorders. Here we characterize mechanisms that regulate trace elements in human cells by designing and performing a genome-wide high-throughput siRNA/ionomics screen, and examining top hits in cellular and biochemical assays. The screen reveals high stability of the ionomes, especially the zinc ionome, and yields known regulators and novel candidates. We further uncover fundamental differences in the regulation of different trace elements. Specifically, selenium levels are controlled through the selenocysteine machinery and expression of abundant selenoproteins; copper balance is affected by lipid metabolism and requires machinery involved in protein trafficking and post-translational modifications; and the iron levels are influenced by iron import and expression of the iron/haeme-containing enzymes. Our approach can be applied to a variety of disease models and/or nutritional conditions, and the generated data set opens new directions for studies of human trace element metabolism. 1 Genetics Division, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts 02115, USA. 2 Department of Biochemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA. 3 Department of Physiology, Johns Hopkins University, Baltimore, Maryland 21205, USA. 4 Key Laboratory of Nutrition and Metabolism, Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Shanghai 200031, China. -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -

The Role of Human Ribosomal Proteins in the Maturation of Rrna and Ribosome Production
JOBNAME: RNA 14#9 2008 PAGE: 1 OUTPUT: Friday August 8 17:34:50 2008 csh/RNA/164293/rna11320 Downloaded from rnajournal.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press The role of human ribosomal proteins in the maturation of rRNA and ribosome production SARA ROBLEDO,1,3 RACHEL A. IDOL,1,3 DAN L. CRIMMINS,2 JACK H. LADENSON,2 PHILIP J. MASON,1,4 and MONICA BESSLER1,4 1Department of Internal Medicine, Division of Hematology, Washington University School of Medicine, St. Louis, Missouri 63110, USA 2Department of Pathology and Immunology, Division of Laboratory and Genomic Medicine, Washington University School of Medicine, St. Louis, Missouri 63110, USA ABSTRACT Production of ribosomes is a fundamental process that occurs in all dividing cells. It is a complex process consisting of the coordinated synthesis and assembly of four ribosomal RNAs (rRNA) with about 80 ribosomal proteins (r-proteins) involving more than 150 nonribosomal proteins and other factors. Diamond Blackfan anemia (DBA) is an inherited red cell aplasia caused by mutations in one of several r-proteins. How defects in r-proteins, essential for proliferation in all cells, lead to a human disease with a specific defect in red cell development is unknown. Here, we investigated the role of r-proteins in ribosome biogenesis in order to find out whether those mutated in DBA have any similarities. We depleted HeLa cells using siRNA for several individual r-proteins of the small (RPS6, RPS7, RPS15, RPS16, RPS17, RPS19, RPS24, RPS25, RPS28) or large subunit (RPL5, RPL7, RPL11, RPL14, RPL26, RPL35a) and studied the effect on rRNA processing and ribosome production. -

Patterns of Ribosomal Protein Expression Specify Normal and Malignant Human Cells Joao C
Guimaraes and Zavolan Genome Biology (2016) 17:236 DOI 10.1186/s13059-016-1104-z RESEARCH Open Access Patterns of ribosomal protein expression specify normal and malignant human cells Joao C. Guimaraes* and Mihaela Zavolan* Abstract Background: Ribosomes are highly conserved molecular machines whose core composition has traditionally been regarded as invariant. However, recent studies have reported intriguing differences in the expression of some ribosomal proteins (RPs) across tissues and highly specific effects on the translation of individual mRNAs. Results: To determine whether RPs are more generally linked to cell identity, we analyze the heterogeneity of RP expression in a large set of human tissues, primary cells, and tumors. We find that about a quarter of human RPs exhibit tissue-specific expression and that primary hematopoietic cells display the most complex patterns of RP expression, likely shaped by context-restricted transcriptional regulators. Strikingly, we uncover patterns of dysregulated expression of individual RPs across cancer types that arise through copy number variations and are predictive for disease progression. Conclusions: Our study reveals an unanticipated plasticity of RP expression across normal and malignant human cell types and provides a foundation for future characterization of cellular behaviors that are orchestrated by specific RPs. Keywords: Ribosomal proteins, Ribosome heterogeneity, Translation, Hematopoiesis, Cancer Background Indeed, many feedback mechanisms have been discov- Protein synthesis is at the core of cellular life. It is car- ered that link the production of different ribosomal ried out by the ribosome, a highly conserved molecular components to maintain an appropriate stoichiometry machine with the same basic architecture in all free- [7]; a number of RPs act in negative feedback loops to living organisms [1–3]. -

Global Analysis of LARP1 Translation Targets Reveals Tunable and Dynamic Features of 5′ TOP Motifs
Global analysis of LARP1 translation targets reveals tunable and dynamic features of 5′ TOP motifs Lucas Philippea,1, Antonia M. G. van den Elzena,1, Maegan J. Watsona, and Carson C. Thoreena,2 aDepartment of Cellular and Molecular Physiology, Yale School of Medicine, New Haven, CT 06510 Edited by Alan G. Hinnebusch, National Institutes of Health, Bethesda, MD, and approved January 29, 2020 (received for review July 25, 2019) Terminal oligopyrimidine (TOP) motifs are sequences at the 5′ ends recent findings have hinted that the RNA-binding protein La- of mRNAs that link their translation to the mTOR Complex 1 related protein 1 (LARP1) may have a central role (8–10). (mTORC1) nutrient-sensing signaling pathway. They are com- LARP1 is a large protein (150 kDa) with several RNA-binding monly regarded as discrete elements that reside on ∼100 mRNAs domains. Its central region contains a La motif (LaM) domain that mostly encode translation factors. However, the full spectrum that defines the La-related protein (LARP) superfamily, along of TOP sequences and their prevalence throughout the transcrip- with an adjacent RNA recognition motif-like (RRM-L) domain. tome remain unclear, primarily because of uncertainty over the Its C terminus encodes a domain known as the DM15 region. This mechanism that detects them. Here, we globally analyzed trans- domain is unique to LARP1 and its closely related homolog lation targets of La-related protein 1 (LARP1), an RNA-binding pro- LARP1B, and is therefore also known as the LARP1 domain (11). tein and mTORC1 effector that has been shown to repress TOP Several observations suggest that LARP1 directly represses TOP mRNA translation in a few specific cases. -

Lncrna SNHG8 Is Identified As a Key Regulator of Acute Myocardial
Zhuo et al. Lipids in Health and Disease (2019) 18:201 https://doi.org/10.1186/s12944-019-1142-0 RESEARCH Open Access LncRNA SNHG8 is identified as a key regulator of acute myocardial infarction by RNA-seq analysis Liu-An Zhuo, Yi-Tao Wen, Yong Wang, Zhi-Fang Liang, Gang Wu, Mei-Dan Nong and Liu Miao* Abstract Background: Long noncoding RNAs (lncRNAs) are involved in numerous physiological functions. However, their mechanisms in acute myocardial infarction (AMI) are not well understood. Methods: We performed an RNA-seq analysis to explore the molecular mechanism of AMI by constructing a lncRNA-miRNA-mRNA axis based on the ceRNA hypothesis. The target microRNA data were used to design a global AMI triple network. Thereafter, a functional enrichment analysis and clustering topological analyses were conducted by using the triple network. The expression of lncRNA SNHG8, SOCS3 and ICAM1 was measured by qRT-PCR. The prognostic values of lncRNA SNHG8, SOCS3 and ICAM1 were evaluated using a receiver operating characteristic (ROC) curve. Results: An AMI lncRNA-miRNA-mRNA network was constructed that included two mRNAs, one miRNA and one lncRNA. After RT-PCR validation of lncRNA SNHG8, SOCS3 and ICAM1 between the AMI and normal samples, only lncRNA SNHG8 had significant diagnostic value for further analysis. The ROC curve showed that SNHG8 presented an AUC of 0.850, while the AUC of SOCS3 was 0.633 and that of ICAM1 was 0.594. After a pairwise comparison, we found that SNHG8 was statistically significant (P SNHG8-ICAM1 = 0.002; P SNHG8-SOCS3 = 0.031). -

Cdna, Genomic Cloning and Sequence Analysis of Ribosomal Protein S4X Gene ( RPS4X ) from Ailuropoda Melanoleuca
African Journal of Biotechnology Vol. 11(21), pp. 4752-4758, 13 March, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB11.3538 ISSN 1684–5315 © 2012 Academic Journals Full Length Research Paper cDNA, genomic cloning and sequence analysis of ribosomal protein S4X gene ( RPS4X ) from Ailuropoda melanoleuca Fu-yan Luo 1, 2 , Wan-ru Hou 2* and Wei-ying Chen 2 1Department of Environmental Science, Sichuan University, No. 24 South Section 1, the First Ring Road, Chengdu, Sichuan 610065, China. 2College of Life Science, China West Normal University, No. 1 Shida Road, Nanchong, Sichuan 637009, China. Accepted 27 February, 2012 Ribosomal protein S4X (RPS4X) is one of the 40S ribosomal proteins encoded by the RPS4X gene. The cDNA and the genomic sequence of RPS4X were cloned successfully from giant panda ( Ailuropoda melanoleuca ) using reverse transcriptase-polymerase chain reaction (RT-PCR) and touchdown-PCR technology respectively, followed by sequencing and analyzing. The cDNA of the RPS4X cloned is 812 bp in size, containing an open reading frame (ORF) of 792 bp encoding 263 amino acids with deduced molecular weight of 29.6 kD and isoelectric point (PI) of 10.16. The length of the genomic sequence is 5074 bp, which was found to possess 7 exons and 6 introns. There are 1 N-glycosylation site, 3 protein kinase C phosphorylation sites, 4 casein kinase II phosphorylation sites, 2 tyrosine kinase phosphorylation sites and 4 N-myristoylation sites in the RPS4X of giant panda by topology prediction. Alignment analysis indicated that the coding sequence and the deduced amino acid sequence of RPS4X gene shows very high homology with other reported mammalian species.