And Copper Homeostatic Mechanisms in Brain Contributes to the Pathogenesis of Neurodegenerative Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Calcium-Dependent Copper Redistributions in Neuronal Cells Revealed by a Fluorescent Copper Sensor and X-Ray Fluorescence Microscopy
Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy Sheel C. Dodania,1, Dylan W. Domaillea,1, Christine I. Nama,b,1, Evan W. Millera, Lydia A. Finneyc, Stefan Vogtc, and Christopher J. Changa,b,2 aDepartment of Chemistry, University of California, Berkeley, CA 94720; bHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; and cX-Ray Sciences Division and Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL 60439 Edited* by Harry B. Gray, California Institute of Technology, Pasadena, CA, and approved February 23, 2011 (received for review July 9, 2010) Dynamic fluxes of s-block metals like potassium, sodium, and Along these lines, molecular imaging with copper-responsive calcium are of broad importance in cell signaling. In contrast, the fluorescent sensors offers a potentially powerful methodology concept of mobile transition metals triggered by cell activation for interrogating its cell biology by allowing the specific tracking remains insufficiently explored, in large part because metals like of copper pools in living cells with spatial and temporal resolution copper and iron are typically studied as static cellular nutrients and (12, 26–32). In this regard, analogous tools have revolutionized there are a lack of direct, selective methods for monitoring their the study of calcium in a variety of biological settings (1) and hold distributions in living cells. To help meet this need, we now report promise for interrogating other cellular metals (26). However, Coppersensor-3 (CS3), a bright small-molecule fluorescent probe fluorescence-based sensing of Cuþ, the oxidation state stabilized that offers the unique capability to image labile copper pools in in reducing cytosolic environments, presents several additional living cells at endogenous, basal levels. -

Current Biomedical Use of Copper Chelation Therapy
International Journal of Molecular Sciences Review Current Biomedical Use of Copper Chelation Therapy Silvia Baldari 1,2, Giuliana Di Rocco 1 and Gabriele Toietta 1,* 1 Department of Research, Advanced Diagnostic, and Technological Innovation, IRCCS Regina Elena National Cancer Institute, via E. Chianesi 53, 00144 Rome, Italy; [email protected] (S.B.); [email protected] (G.D.R.) 2 Department of Medical Surgical Sciences and Biotechnologies, University of Rome “La Sapienza”, C.so della Repubblica 79, 04100 Latina, Italy * Correspondence: [email protected]; Tel.: +39-06-5266-2604 Received: 9 January 2020; Accepted: 4 February 2020; Published: 6 February 2020 Abstract: Copper is an essential microelement that plays an important role in a wide variety of biological processes. Copper concentration has to be finely regulated, as any imbalance in its homeostasis can induce abnormalities. In particular, excess copper plays an important role in the etiopathogenesis of the genetic disease Wilson’s syndrome, in neurological and neurodegenerative pathologies such as Alzheimer’s and Parkinson’s diseases, in idiopathic pulmonary fibrosis, in diabetes, and in several forms of cancer. Copper chelating agents are among the most promising tools to keep copper concentration at physiological levels. In this review, we focus on the most relevant compounds experimentally and clinically evaluated for their ability to counteract copper homeostasis deregulation. In particular, we provide a general overview of the main disorders characterized by a pathological increase in copper levels, summarizing the principal copper chelating therapies adopted in clinical trials. Keywords: copper; chelation therapy; therapeutic chelation; metal homeostasis; cancer; metalloproteins 1. -

Ncomms4301.Pdf
ARTICLE Received 8 Jul 2013 | Accepted 23 Jan 2014 | Published 13 Feb 2014 DOI: 10.1038/ncomms4301 Genome-wide RNAi ionomics screen reveals new genes and regulation of human trace element metabolism Mikalai Malinouski1,2, Nesrin M. Hasan3, Yan Zhang1,4, Javier Seravalli2, Jie Lin4,5, Andrei Avanesov1, Svetlana Lutsenko3 & Vadim N. Gladyshev1 Trace elements are essential for human metabolism and dysregulation of their homoeostasis is associated with numerous disorders. Here we characterize mechanisms that regulate trace elements in human cells by designing and performing a genome-wide high-throughput siRNA/ionomics screen, and examining top hits in cellular and biochemical assays. The screen reveals high stability of the ionomes, especially the zinc ionome, and yields known regulators and novel candidates. We further uncover fundamental differences in the regulation of different trace elements. Specifically, selenium levels are controlled through the selenocysteine machinery and expression of abundant selenoproteins; copper balance is affected by lipid metabolism and requires machinery involved in protein trafficking and post-translational modifications; and the iron levels are influenced by iron import and expression of the iron/haeme-containing enzymes. Our approach can be applied to a variety of disease models and/or nutritional conditions, and the generated data set opens new directions for studies of human trace element metabolism. 1 Genetics Division, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts 02115, USA. 2 Department of Biochemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA. 3 Department of Physiology, Johns Hopkins University, Baltimore, Maryland 21205, USA. 4 Key Laboratory of Nutrition and Metabolism, Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Shanghai 200031, China. -

Essential Trace Elements in Human Health: a Physician's View
Margarita G. Skalnaya, Anatoly V. Skalny ESSENTIAL TRACE ELEMENTS IN HUMAN HEALTH: A PHYSICIAN'S VIEW Reviewers: Philippe Collery, M.D., Ph.D. Ivan V. Radysh, M.D., Ph.D., D.Sc. Tomsk Publishing House of Tomsk State University 2018 2 Essential trace elements in human health UDK 612:577.1 LBC 52.57 S66 Skalnaya Margarita G., Skalny Anatoly V. S66 Essential trace elements in human health: a physician's view. – Tomsk : Publishing House of Tomsk State University, 2018. – 224 p. ISBN 978-5-94621-683-8 Disturbances in trace element homeostasis may result in the development of pathologic states and diseases. The most characteristic patterns of a modern human being are deficiency of essential and excess of toxic trace elements. Such a deficiency frequently occurs due to insufficient trace element content in diets or increased requirements of an organism. All these changes of trace element homeostasis form an individual trace element portrait of a person. Consequently, impaired balance of every trace element should be analyzed in the view of other patterns of trace element portrait. Only personalized approach to diagnosis can meet these requirements and result in successful treatment. Effective management and timely diagnosis of trace element deficiency and toxicity may occur only in the case of adequate assessment of trace element status of every individual based on recent data on trace element metabolism. Therefore, the most recent basic data on participation of essential trace elements in physiological processes, metabolism, routes and volumes of entering to the body, relation to various diseases, medical applications with a special focus on iron (Fe), copper (Cu), manganese (Mn), zinc (Zn), selenium (Se), iodine (I), cobalt (Co), chromium, and molybdenum (Mo) are reviewed. -

Supplementary Material and Methods
Supplementary material and methods Generation of cultured human epidermal sheets Normal human epidermal keratinocytes were isolated from human breast skin. Keratinocytes were grown on a feeder layer of irradiated human fibroblasts pre-seeded at 4000 cells /cm² in keratinocyte culture medium (KCM) containing a mix of 3:1 DMEM and HAM’s F12 (Invitrogen, Carlsbad, USA), supplemented with 10% FCS, 10ng/ml epidermal growth factor (EGF; R&D systems, Minneapolis, MN, USA), 0.12 IU/ml insulin (Lilly, Saint- Cloud, France), 0.4 mg/ml hydrocortisone (UpJohn, St Quentin en Yvelelines, France) , 5 mg/ml triiodo-L- thyronine (Sigma, St Quentin Fallavier, France), 24.3 mg/ml adenine (Sigma), isoproterenol (Isuprel, Hospira France, Meudon, France) and antibiotics (20 mg/ml gentamicin (Phanpharma, Fougères, France), 100 IU/ml penicillin (Phanpharma), and 1 mg/ml amphotericin B (Phanpharma)). The medium was changed every two days. NHEK were then cultured over a period of 13 days according to the protocol currently used at the Bank of Tissues and Cells for the generation of clinical grade epidermal sheets used for the treatment of severe extended burns (Ref). When needed, cells were harvested with trypsin-EDTA 0.05% (Thermo Fisher Scientific, Waltham, MA, USA) and collected for analysis. Clonogenic assay Keratinocytes were seeded on a feeder layer of irradiated fibroblasts, at a clonal density of 10-20 cells/cm² and cultivated for 10 to 14 days. Three flasks per tested condition were fixed and colored in a single 30 mns step using rhodamine B (Sigma) diluted at 0.01 g/ml in 4% paraformaldehyde. In each tested condition, cells from 3 other flasks were numerated after detachment by trypsin treatment. -

Effect of Dietary Copper Deficiency on Iron Metabolism in the Pregnant
Downloaded from British Journal of Nutrition (2007), 97, 239–246 DOI: 10.1017/S0007114507239960 q The Authors 2007 https://www.cambridge.org/core Effect of dietary copper deficiency on iron metabolism in the pregnant rat Henriette S. Andersen1, Lorraine Gambling1, Grietje Holtrop2 and Harry J. McArdle1* 1 Rowett Research Institute, Greenburn Road, Bucksburn, Aberdeen AB21 9SB, UK . IP address: 2BioSS, Rowett Research Institute, Greenburn Road, Bucksburn, Aberdeen AB21 9SB, UK (Received 13 June 2006 – Revised 4 September 2006 – Accepted 6 September 2006) 170.106.33.14 Cu and Fe metabolism are known to be linked, but the interactions during pregnancy are less well studied. In the present study we used rats to examine the effect of Cu deficiency during pregnancy on Fe and Cu levels in maternal and fetal tissue and on the gene expression profile of , on proteins involved in Cu and Fe metabolism in the placenta. Rats were fed diets with different Cu contents before and during pregnancy. Samples 28 Sep 2021 at 13:17:06 were collected on day 21 of gestation. Cu levels, ceruloplasmin activity and serum Fe all decreased in maternal serum of Cu-deficient animals. Maternal liver Fe inversely correlated with liver Cu. Placental Cu levels decreased with no change in Fe. Fe and Cu levels both decreased in the fetal liver. The drop in maternal liver Cu was significantly correlated with a decrease in organ weight of fetal liver, lung and kidney. No changes were observed in mRNA expression of Cu transporter 1, Menkes P-type Cu-ATPase 7A, Wilson P-type Cu-ATPase 7B, cytochrome-c oxidase, and Cu chaperone Atox1 in the placenta of Cu-deficient dams. -

ATOX1 (NM 004045) Human Untagged Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC117628 ATOX1 (NM_004045) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: ATOX1 (NM_004045) Human Untagged Clone Tag: Tag Free Symbol: ATOX1 Synonyms: ATX1; HAH1 Vector: pCMV6-XL4 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >OriGene ORF within SC117628 sequence for NM_004045 edited (data generated by NextGen Sequencing) ATGCCGAAGCACGAGTTCTCTGTGGACATGACCTGTGGAGGCTGTGCTGAAGCTGTCTCT CGGGTCCTCAATAAGCTTGGAGGAGTTAAGTATGACATTGACCTGCCCAACAAGAAGGTC TGCATTGAATCTGAGCACAGCATGGACACTCTGCTTGCAACCCTGAAGAAAACAGGAAAG ACTGTTTCCTACCTTGGCCTTGAGTAG Clone variation with respect to NM_004045.3 5' Read Nucleotide >OriGene 5' read for NM_004045 unedited Sequence: GTAACGTCAGAATTTGTATACGACTCACTATAGGGCGGCCGCGAATTCGCACCAGCACCG CCGCCACACCGCCGCCACACCGCCGCTGCCTCAGTCATGCCGAAGCACGAGTTCTCTGTG GACATGACCTGTGGAGGCTGTGCTGAAGCTGTCTCTCGGGTCCTCAATAAGCTTGGAGGA GTTAAGTATGACATTGACCTGCCCAACAAGAAGGTCTGCATTGAATCTGAGCACAGCATG GACACTCTGCTTGCAACCCTGAAGAAAACAGGAAAGACTGTTTCCTACCTTGGCCTTGAG TAGCAGGGGCCTGGTCCCCACAGCCCACAGGATGGACCAAAGGGGGCAGGATGCTGATCC TCCCGCTGGCTTCCAGACAGACCTGGGACTTGGCAGTCATGCCGGGTGATGGTGTTCCTG CGGAGACCCTCAGTTGTCCTATTCCTTCCTAGCTTCCCTGCAATAAAATCAAGCTGCTTT TGTTGGNAAANAAAAAAAANNNNNAAAAAAAAAAAAAAAAAAAAAAAAANAAAAAAAAAA AAAAAAAAAAAAAAAAAAACCCTCGACTTTAGATTGCGGCCGCGGTCATAGCTGTTTCCT GAACAGATCCCGGGTGGCATCCCTGTGACCCCTCCCAAGTGCCTCTCCTGGCCCTGAAGG -

Transcriptomic and Functional Analysis of Aβ1-42 Oligomer-Stimulated
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.12.456055; this version posted August 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Transcriptomic and functional analysis of A1-42 oligomer-stimulated human monocyte-derived microglia-like cells Tamar Smit1,2, Paul R. Ormel1, Jacqueline A. Sluijs1, Lianne A. Hulshof1, Jinte Middeldorp1,3, Lot D. de Witte4, Elly M. Hol1*, Vanessa Donega1* 1Department of Translational Neuroscience, Brain Center, University Medical Center Utrecht, Utrecht University, 3584 CG Utrecht, The Netherlands 2Swammerdam Institute for Life Sciences, Center for Neuroscience, University of Amsterdam, 1098 XH Amsterdam, The Netherlands 3Department of Immunobiology, Biomedical Primate Research Centre, Rijswijk, The Netherlands 4Department of Psychiatry, Icahn School of medicine at Mount Sinai, New York, NY, USA *Authors contributed equally to this work Corresponding author: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.08.12.456055; this version posted August 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Dysregulation of microglial function contributes to Alzheimer’s disease (AD) pathogenesis. Several genetic and transcriptome studies have revealed microglia specific genetic risk factors, and changes in microglia expression profiles in AD pathogenesis, viz. -

The Bioinorganic Periodic Table
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2019 The Bioinorganic Periodic Table Freisinger, Eva ; Sigel, Roland K O Abstract: Life depends on metals. While carbon, in terms of abundance and versatility, is considered THE element of life, the vast variety and diversity of the chemistry taking place in living organisms could not be achieved without metal ions. More than twenty metals are found in the human body, most of them being essential, some beneficial, and for others it is still unknown what role they might fulfilina living cell. Here we give a short introduction into the bioinorganic world of the periodic table, providing just a few examples of key metals for life and aiming to give a flavour to gain further insights into this exciting field of inorganic chemistry at the intersection to the life sciences. DOI: https://doi.org/10.2533/chimia.2019.185 Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-170342 Journal Article Published Version Originally published at: Freisinger, Eva; Sigel, Roland K O (2019). The Bioinorganic Periodic Table. CHIMIA International Journal for Chemistry, 73(3):185-193. DOI: https://doi.org/10.2533/chimia.2019.185 InternatIonal Year of the PerIodIc table CHIMIA 2019, 73, No. 3 185 doi:10.2533/chimia.2019.185 Chimia 73 (2019) 185–193 © Swiss Chemical Society The Bioinorganic Periodic Table Eva Freisinger* and Roland K. O. Sigel* Abstract: Life depends on metals. While carbon, in terms of abundance and versatility, is considered THE ele- ment of life, the vast variety and diversity of the chemistry taking place in living organisms could not be achieved without metal ions. -

Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared Amongst Δγ T, Innate Lymphoid, and Th Cells
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 δγ is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2016; 197:1460-1470; Prepublished online 6 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600643 http://www.jimmunol.org/content/197/4/1460 Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst T, Innate Lymphoid, and Th Cells You Jeong Lee, Gabriel J. Starrett, Seungeun Thera Lee, Rendong Yang, Christine M. Henzler, Stephen C. Jameson and Kristin A. Hogquist J Immunol cites 41 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2016/07/06/jimmunol.160064 3.DCSupplemental This article http://www.jimmunol.org/content/197/4/1460.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. The Journal of Immunology Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gd T, Innate Lymphoid, and Th Cells You Jeong Lee,* Gabriel J. -
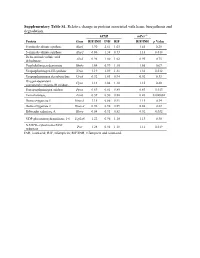
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-