European Sea Bass Genome and Its Variation Provide Insights Into Adaptation to Euryhalinity and Speciation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

Genome-Wide Association Identifies Candidate Genes That Influence The
Genome-wide association identifies candidate genes that influence the human electroencephalogram Colin A. Hodgkinsona,1, Mary-Anne Enocha, Vibhuti Srivastavaa, Justine S. Cummins-Omana, Cherisse Ferriera, Polina Iarikovaa, Sriram Sankararamanb, Goli Yaminia, Qiaoping Yuana, Zhifeng Zhoua, Bernard Albaughc, Kenneth V. Whitea, Pei-Hong Shena, and David Goldmana aLaboratory of Neurogenetics, National Institute on Alcohol Abuse and Alcoholism, Rockville, MD 20852; bComputer Science Department, University of California, Berkeley, CA 94720; and cCenter for Human Behavior Studies, Weatherford, OK 73096 Edited* by Raymond L. White, University of California, Emeryville, CA, and approved March 31, 2010 (received for review July 23, 2009) Complex psychiatric disorders are resistant to whole-genome reflects rhythmic electrical activity of the brain. EEG patterns analysis due to genetic and etiological heterogeneity. Variation in dynamically and quantitatively index cortical activation, cognitive resting electroencephalogram (EEG) is associated with common, function, and state of consciousness. EEG traits were among the complex psychiatric diseases including alcoholism, schizophrenia, original intermediate phenotypes in neuropsychiatry, having been and anxiety disorders, although not diagnostic for any of them. EEG first recorded in humans in 1924 by Hans Berger, who documented traits for an individual are stable, variable between individuals, and the α rhythm, seen maximally during states of relaxation with eyes moderately to highly heritable. Such intermediate phenotypes closed, and supplanted by faster β waves during mental activity. appear to be closer to underlying molecular processes than are EEG can be used clinically for the evaluation and differential di- clinical symptoms, and represent an alternative approach for the agnosis of epilepsy and sleep disorders, differentiation of en- identification of genetic variation that underlies complex psychiat- cephalopathy from catatonia, assessment of depth of anesthesia, ric disorders. -

Gene Expression Is Related to Parental Origin and Regional Coordinate Control
Journal of Human Genetics (2009) 54, 193–198 & 2009 The Japan Society of Human Genetics All rights reserved 1434-5161/09 $32.00 www.nature.com/jhg ORIGINAL ARTICLE William’s syndrome: gene expression is related to parental origin and regional coordinate control Jeremy C Collette1, Xiao-Ning Chen1, Debra L Mills2, Albert M Galaburda3, Allan L Reiss4, Ursula Bellugi5 and Julie R Korenberg1,6 William’s syndrome (WS) features a spectrum of neurocognitive and behavioral abnormalities due to a rare 1.5 MB deletion that includes about 24–28 genes on chromosome band 7q11.23. Study of the expression of these genes from the single normal copy provides an opportunity to elucidate the genetic and epigenetic controls on these genes as well as their roles in both WS and normal brain development and function. We used quantitative RT-PCR to determine the transcriptional level of 14 WS gene markers in a cohort of 77 persons with WS and 48 normal controls. Results reported here: (1) show that the expression of the genes deleted in WS is decreased in some but not all cases, (2) demonstrate that the parental origin of the deletion contributes to the level of expression of GTF2I independently of age and gender and (3) indicate that the correlation of expression between GTF2I and some other genes in the WS region differs in WS subjects and normal controls, which in turn points toward a regulatory role for this gene. Interspecies comparisons suggest GTF2I may play a key role in normal brain development. Journal of Human Genetics (2009) 54, 193–198; doi:10.1038/jhg.2009.5; published online 13 March 2009 Keywords: William’s syndrome; gene expression; RT-PCR; parental origin; GTF2I INTRODUCTION As an approach toward understanding the role of the deleted genes William’s syndrome (WS) is a neurogenetic disorder affecting human in WS, we have characterized WS subjects according to genetic, social/ development and adult cognition. -
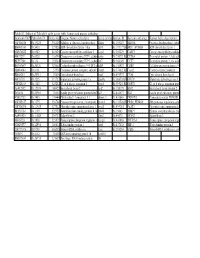
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Predicting Gene Ontology Biological Process from Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R
Methods Predicting Gene Ontology Biological Process From Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R. Hvidsten,2 Herman Midelfart,2 Jan Komorowski,2,3,4 and Arne K. Sandvik1 1Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, N-7489 Trondheim, Norway; 2Department of Information and Computer Science, Norwegian University of Science and Technology, N-7491 Trondheim, Norway; 3The Linnaeus Centre for Bioinformatics, Uppsala University, SE-751 24 Uppsala, Sweden The aim of the present study was to generate hypotheses on the involvement of uncharacterized genes in biological processes. To this end,supervised learning was used to analyz e microarray-derived time-series gene expression data. Our method was objectively evaluated on known genes using cross-validation and provided high-precision Gene Ontology biological process classifications for 211 of the 213 uncharacterized genes in the data set used. In addition,new roles in biological process were hypothesi zed for known genes. Our method uses biological knowledge expressed by Gene Ontology and generates a rule model associating this knowledge with minimal characteristic features of temporal gene expression profiles. This model allows learning and classification of multiple biological process roles for each gene and can predict participation of genes in a biological process even though the genes of this class exhibit a wide variety of gene expression profiles including inverse coregulation. A considerable number of the hypothesized new roles for known genes were confirmed by literature search. In addition,many biological process roles hypothesi zed for uncharacterized genes were found to agree with assumptions based on homology information. -

Role of Coiled-Coil Registry Shifts in Activation of Human Bicaudal D2 for Dynein Recruitment Upon Cargo-Binding
bioRxiv preprint doi: https://doi.org/10.1101/638536; this version posted May 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Role of coiled-coil registry shifts in activation of human Bicaudal D2 for dynein recruitment upon cargo-binding Crystal R. Noell,1,4 Jia Ying Loh, 1,4 Erik W. Debler,2 Kyle M. Loftus,1 Heying Cui,1 Blaine B. Russ,1 Puja Goyal,1,* Sozanne R. Solmaz1,3,* 1Department of Chemistry, State University of New York at Binghamton, Binghamton, NY 13902 2Department of Biochemistry & Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107. 3Lead contact. 4First author equally contributed *To whom the correspondence should be addressed: Sozanne R Solmaz Department of Chemistry, State University of New York at Binghamton PO Box 6000 Binghamton NY 13902 [email protected] +1 607 777 2089 Puja Goyal Department of Chemistry, State University of New York at Binghamton PO Box 6000 Binghamton NY 13902 [email protected] +1 607 777 4308 1 bioRxiv preprint doi: https://doi.org/10.1101/638536; this version posted May 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Highlights • Stable, bona fide BicD2 coiled-coils with distinct registries can be formed. • We provide evidence that a human disease mutation causes a coiled-coil registry shift. • A coiled-coil registry shift could relieve BicD2-autoinhibition upon cargo-binding. • The ability to undergo registry shifts may be an inherent property of coiled-coils. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

RNA Editing at Baseline and Following Endoplasmic Reticulum Stress
RNA Editing at Baseline and Following Endoplasmic Reticulum Stress By Allison Leigh Richards A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Human Genetics) in The University of Michigan 2015 Doctoral Committee: Professor Vivian G. Cheung, Chair Assistant Professor Santhi K. Ganesh Professor David Ginsburg Professor Daniel J. Klionsky Dedication To my father, mother, and Matt without whom I would never have made it ii Acknowledgements Thank you first and foremost to my dissertation mentor, Dr. Vivian Cheung. I have learned so much from you over the past several years including presentation skills such as never sighing and never saying “as you can see…” You have taught me how to think outside the box and how to create and explain my story to others. I would not be where I am today without your help and guidance. Thank you to the members of my dissertation committee (Drs. Santhi Ganesh, David Ginsburg and Daniel Klionsky) for all of your advice and support. I would also like to thank the entire Human Genetics Program, and especially JoAnn Sekiguchi and Karen Grahl, for welcoming me to the University of Michigan and making my transition so much easier. Thank you to Michael Boehnke and the Genome Science Training Program for supporting my work. A very special thank you to all of the members of the Cheung lab, past and present. Thank you to Xiaorong Wang for all of your help from the bench to advice on my career. Thank you to Zhengwei Zhu who has helped me immensely throughout my thesis even through my panic. -

Mammalian Protein Expression and Characterization Tools for Next Generation Biologics
Mammalian protein expression and characterization tools for next generation biologics next generation for tools characterization and expression protein Mammalian Doctoral Thesis in Biotechnology Mammalian protein expression and characterization tools for next generation biologics NIKLAS THALÉN ISBN TRITACBHFOU: KTH www.kth.se Stockholm, Sweden Mammalian protein expression and characterization tools for next generation biologics NIKLAS THALÉN Academic Dissertation which, with due permission of the KTH Royal Institute of Technology, is submitted for public defence for the Degree of Doctor of Philosophy on June 11th, 2021 at 10:00, F3, Lindstedsvägen 26, våningsplan 2, Sing-Sing, KTH campus, Stockholm. Doctoral Thesis in Biotechnology KTH Royal Institute of Technology Stockholm, Sweden 2021 © Niklas Thalén ISBN 978-91-7873-927-1 TRITA-CBH-FOU-2021:21 Printed by: Universitetsservice US-AB, Sweden 2021 Till Sofia Abstract Protein therapeutics are increasingly important for modern medicine. Novel recombinant proteins developed today can bind towards their target with high specificity and with low adverse effect. This has enabled the treatment of diseases that for a few years ago were deemed uncurable. Discovery of therapeutic proteins is driven through protein engineering, a field that is in constant expansion. And, through artificial construction of recombinant proteins, a large array of diseases can be defeated. The function and quality of these protein therapeutics rely on the correct folding, assembly and residue modification that occurs during their production within a living production cell host. Furthermore, producing them in large quantities are essential for accessibility of the best biopharmaceuticals available. Commonly, mammalian cells are the production host of choice when it comes to production of biopharmaceuticals. -

Salinity Regulates Claudin Mrna and Protein Expression in the Teleost Gill
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Am J Physiol Regul Integr Comp Physiol 294: R1004–R1014, 2008. provided by University of Southern Denmark Research Output First published January 9, 2008; doi:10.1152/ajpregu.00112.2007. Salinity regulates claudin mRNA and protein expression in the teleost gill Christian K. Tipsmark,* David A. Baltzegar,* Ozkan Ozden, Brenda J. Grubb, and Russell J. Borski Department of Zoology, North Carolina State University, Raleigh, North Carolina Submitted 14 February 2007; accepted in final form 19 December 2007 Tipsmark CK, Baltzegar DA, Ozden O, Grubb BJ, Borski RJ. The biochemical basis for changes in water permeability may Salinity regulates claudin mRNA and protein expression in the teleost entail the robust downregulation of gill aquaporin mRNA and gill. Am J Physiol Regul Integr Comp Physiol 294: R1004–R1014, 2008. protein expression that accompanies SW acclimation in eel (6, First published January 9, 2008; doi:10.1152/ajpregu.00112.2007.—The 21). Although osmotic permeability decreases in SW, the teleost gill carries out NaCl uptake in freshwater (FW) and NaCl conductance and short-circuit current is found to be larger in a excretion in seawater (SW). This transformation with salinity requires close regulation of ion transporter capacity and epithelial permeabil- SW teleost compared with a FW fish (8, 27). A simultaneous change in the ultrastructure of tight junctions (TJs) is also seen Downloaded from ity. This study investigates the regulation of tight-junctional claudins ϩ during salinity acclimation in fish. We identified claudin 3- and (28). Secretion of Na is thought to involve a paracellular claudin 4-like immunoreactive proteins and examined their expression path confined to thin “leaky” TJs that occur in SW between and that of select ion transporters by performing Western blot in mature chloride cells and accessory cells (19, 36), and this tilapia (Oreochromis mossambicus) gill during FW and SW acclima- may contribute to the relatively high ionic permeability of tion. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

CCT6A Sirna (Mouse)
For research purposes only, not for human use Product Data Sheet CCT6A siRNA (Mouse) Catalog # Source Reactivity Applications CRM0566 Synthetic M RNAi Description siRNA to inhibit CCT6A expression using RNA interference Specificity CCT6A siRNA (Mouse) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol CCT6A Alternative Names CCT6; CCTZ; CCTZ1; T-complex protein 1 subunit zeta; TCP-1-zeta; CCT-zeta-1 Entrez Gene 12466 (Mouse) SwissProt P80317 (Mouse) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of mouse CCT6A gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB-