The USH2A Gene: an Analysis of Ultrasonic Vocalizations in a Mouse Model of Usher Syndrome Type 2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Genetic Heterogeneity of Usher Syndrome Type II
J7Med Genet 1996;33:753-757 753 Genetic heterogeneity of Usher syndrome type II in a Dutch population J Med Genet: first published as 10.1136/jmg.33.9.753 on 1 September 1996. Downloaded from S Pieke-Dahl, A van Aarem, A Dobin, C W R J Cremers, W J Kimberling Abstract In 1959, Hallgren' laid the foundation for the The Usher syndromes are a group ofauto- clinical definition of US in a study of 172 US somal recessive disorders characterised patients from Sweden. Hallgren' showed sig- by retinitis pigmentosa (RP) with con- nificant phenotypic heterogeneity of US by genital, stable (non-progressive) sen- describing two clinically distinct forms, Usher sorineural hearing loss. Profound deaf- syndrome type I and Usher syndrome type II. ness, RP, and no vestibular responses are Profound congenital deafness, RP, and absent features of Usher type I, whereas moder- vestibular responses was defined as Usher I, ate to severe hearing loss and RP with while those exhibiting a congenital moderate to normal vestibular function describe severe hearing loss, RP, and no associated ves- Usher type II. The gene responsible for tibular problems was defined as Usher II.2 3 most cases ofUsher II, USH2a, is on chro- Although the existence of a type of US with mosome 1q41; at least one other Usher II progressive hearing loss had been proposed,4 gene (as yet unlinked) is known to exist. there was no firm genetic evidence for a Usher III presents with a progressive separate Usher III phenotype until a group of hearing loss that can mimic the audiomet- Finnish families with a phenotype of progres- ric profile seen in Usher II. -

The USH2A C.2299Delg Mutation: Dating Its Common Origin in a Southern European Population
European Journal of Human Genetics (2010) 18, 788–793 & 2010 Macmillan Publishers Limited All rights reserved 1018-4813/10 www.nature.com/ejhg ARTICLE The USH2A c.2299delG mutation: dating its common origin in a Southern European population Elena Aller1,2, Lise Larrieu3, Teresa Jaijo1,2, David Baux3, Carmen Espino´s2, Fernando Gonza´lez-Candelas4,5,6, Carmen Na´jera7, Francesc Palau2,8, Mireille Claustres3,9,10, Anne-Franc¸oise Roux3,9 and Jose´ M Milla´n*,1,2 Usher syndrome type II is the most common form of Usher syndrome. USH2A is the main responsible gene of the three known to be disease causing. It encodes two isoforms of the protein usherin. This protein is part of an interactome that has an essential role in the development and function of inner ear hair cells and photoreceptors. The gene contains 72 exons spanning over a region of 800 kb. Although numerous mutations have been described, the c.2299delG mutation is the most prevalent in several populations. Its ancestral origin was previously suggested after the identification of a common core haplotype restricted to 250 kb in the 5¢ region that encodes the short usherin isoform. By extending the haplotype analysis over the 800 kb region of the USH2A gene with a total of 14 intragenic single nucleotide polymorphisms, we have been able to define 10 different c.2299delG haplotypes, showing high variability but preserving the previously described core haplotype. An exhaustive c.2299delG/control haplotype study suggests that the major source of variability in the USH2A gene is recombination. Furthermore, we have evidenced twice the amount of recombination hotspots located in the 500 kb region that covers the 3¢ end of the gene, explaining the higher variability observed in this region when compared with the 250 kb of the 5¢ region. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Mutation Screening of the USH2A Gene in Retinitis Pigmentosa and USHER Patients in a Han Chinese Population
Eye (2018) 32:1608–1614 https://doi.org/10.1038/s41433-018-0130-3 ARTICLE Mutation screening of the USH2A gene in retinitis pigmentosa and USHER patients in a Han Chinese population 1,2,3 1 1 1 4 1,2,3 Lulin Huang ● Yao Mao ● Jiyun Yang ● Yuanfeng Li ● Yang Li ● Zhenglin Yang Received: 12 October 2016 / Revised: 1 January 2018 / Accepted: 25 April 2018 / Published online: 13 June 2018 © The Author(s) 2018. This article is published with open access Abstract Objectives USH2A encodes for usherin, a basement membrane protein in the inner ear and retina. USH2A can cause retinitis pigmentosa (RP) with or without hearing loss. The aim of this study was to detect USH2A mutations in a Chinese cohort of 75 small RP families and 10 Usher syndrome families. Methods We performed a direct Sanger sequencing analysis of the USH2A gene to identify mutations for this cohort. Results We identified a total of eight mutations in four of the 75 small RP families (5.3%) and two mutations in one of the 10 Usher families (10%); all families were detected to have compound heterozygous mutations. In families with non- syndromic RP, we identified the compound heterozygous mutations p.Pro4818Leuand p.Leu2395Hisfs*19 in family No. 1234567890();,: 1234567890();,: 19114, p.Arg4493His and p.His1677Glnfs*15 in family No.19162, c.8559-2A > G and p.Arg1549* in family No.19123 and p.Ser5060Pro and p.Arg34Leufs*41 in family No.19178. We also identified the heterozygous mutations p.Arg3719His and p.Cys934Trp in family No.19124, which was the Usher syndrome family. -

Mouse Mutants As Models for Congenital Retinal Disorders
Experimental Eye Research 81 (2005) 503–512 www.elsevier.com/locate/yexer Review Mouse mutants as models for congenital retinal disorders Claudia Dalke*, Jochen Graw GSF-National Research Center for Environment and Health, Institute of Developmental Genetics, D-85764 Neuherberg, Germany Received 1 February 2005; accepted in revised form 1 June 2005 Available online 18 July 2005 Abstract Animal models provide a valuable tool for investigating the genetic basis and the pathophysiology of human diseases, and to evaluate therapeutic treatments. To study congenital retinal disorders, mouse mutants have become the most important model organism. Here we review some mouse models, which are related to hereditary disorders (mostly congenital) including retinitis pigmentosa, Leber’s congenital amaurosis, macular disorders and optic atrophy. q 2005 Elsevier Ltd. All rights reserved. Keywords: animal model; retina; mouse; gene mutation; retinal degeneration 1. Introduction Although mouse models are a good tool to investigate retinal disorders, one should keep in mind that the mouse Mice suffering from hereditary eye defects (and in retina is somehow different from a human retina, particular from retinal degenerations) have been collected particularly with respect to the number and distribution of since decades (Keeler, 1924). They allow the study of the photoreceptor cells. The mouse as a nocturnal animal molecular and histological development of retinal degener- has a retina dominated by rods; in contrast, cones are small ations and to characterize the genetic basis underlying in size and represent only 3–5% of the photoreceptors. Mice retinal dysfunction and degeneration. The recent progress of do not form cone-rich areas like the human fovea. -

Functional Annotation of a Full-Length Mouse Cdna Collection
articles Functional annotation of a full-length mouse cDNA collection The RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium* ...............................................................................................................................* A full list of authors appears at the end of the paper ............................................................................................................................................. The RIKEN Mouse Gene Encyclopaedia Project, a systematic approach to determining the full coding potential of the mouse genome, involves collection and sequencing of full-length complementary DNAs and physical mapping of the corresponding genes to the mouse genome. We organized an international functional annotation meeting (FANTOM) to annotate the ®rst 21,076 cDNAs to be analysed in this project. Here we describe the ®rst RIKEN clone collection, which is one of the largest described for any organism. Analysis of these cDNAs extends known gene families and identi®es new ones. In mammals and higher plants, interpreting the genome sequence is Annotation of cDNAs not straightforward: coding regions are interspersed with noncod- An international meeting was held to facilitate functional annota- ing DNA, and an individual gene may give rise to many gene tion of the cDNA sequences. Participants contributed to the devel- products. Thus, genomic sequence cannot be reliably decoded to opment of a web-based annotation interface that should expedite identify the spectrum of messenger RNAs (the transcriptome) and future annotation of additional clones in the Mouse Gene Encyclo- their corresponding protein products (the proteome). This problem paedia project. We agreed on annotation vocabularies and the is illustrated by the different estimates of the number of human application of Gene Ontology (GO) terms (http://genome.gsc. genes (30,000, 35,000 and 120,000)1±3. -

Usherin Is Required for Maintenance of Retinal Photoreceptors and Normal Development of Cochlear Hair Cells
Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells Xiaoqing Liu*, Oleg V. Bulgakov*, Keith N. Darrow†, Basil Pawlyk*, Michael Adamian*, M. Charles Liberman†, and Tiansen Li*‡ *Berman–Gund Laboratory for the Study of Retinal Degenerations and †Eaton–Peabody Laboratory, Harvard Medical School, Massachusetts Eye and Ear Infirmary, Boston, MA 02114 Edited by Jeremy Nathans, Johns Hopkins University School of Medicine, Baltimore, MD, and approved January 18, 2007 (received for review December 11, 2006) Usher syndrome type IIA (USH2A), characterized by progressive pho- USH2A gene in the human genome, expanding the length of coding toreceptor degeneration and congenital moderate hearing loss, is the sequence to 15 kb (15), encoding a putative 600-kDa protein. In most common subtype of Usher syndrome. In this article, we show addition to the greatly expanded size, the recently identified exons that the USH2A protein, also known as usherin, is an exceptionally also were predicted to encode a membrane-spanning segment large (Ϸ600-kDa) matrix protein expressed specifically in retinal followed by an intracellular PDZ-binding domain at the C terminus. photoreceptors and developing cochlear hair cells. In mammalian There have been conflicting reports about usherin tissue distri- photoreceptors, usherin is localized to a spatially restricted mem- bution and subcellular localization (4, 13, 16–18). The putative brane microdomain at the apical inner segment recess that wraps 600-kDa full-length protein never was confirmed from native around the connecting cilia, corresponding to the periciliary ridge tissues, and the function of usherin was poorly understood. It is complex described for amphibian photoreceptors. -

USH2A Gene Usherin
USH2A gene usherin Normal Function The USH2A gene provides instructions for making a protein called usherin. Usherin is an important component of basement membranes, which are thin, sheet-like structures that separate and support cells in many tissues. Usherin is found in basement membranes in the inner ear and in the retina, which is the layer of light-sensitive tissue at the back of the eye. Although the function of usherin has not been well established, studies suggest that it is part of a group of proteins (a protein complex) that plays an important role in the development and maintenance of cells in the inner ear and retina. The protein complex may also be involved in the function of synapses, which are junctions between nerve cells where cell-to-cell communication occurs. Health Conditions Related to Genetic Changes Retinitis pigmentosa Several dozen mutations in the USH2A gene have been reported to cause retinitis pigmentosa, a vision disorder that causes the light-sensing cells of the retina to gradually deteriorate. USH2A gene mutations are the most common cause of the autosomal recessive form of retinitis pigmentosa, accounting for 10 to 15 percent of all cases. This form of the disorder is described as nonsyndromic, which means that it is not associated with other signs and symptoms as part of a genetic syndrome (such as Usher syndrome, described below). The USH2A gene mutations that cause retinitis pigmentosa change single protein building blocks (amino acids) in the usherin protein. Through a mechanism that is not well understood, these genetic changes lead to the gradual breakdown of specialized light receptor cells called photoreceptors in the retina. -

Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients
International Journal of Molecular Sciences Article Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients Akira Inaba 1,2,3, Akiko Maeda 1,2,*, Akiko Yoshida 1,2, Kanako Kawai 1,2, Yasuhiko Hirami 1,2, Yasuo Kurimoto 1,2, Shinji Kosugi 3 and Masayo Takahashi 1,2 1 Department of Ophthalmology, Kobe City Eye Hospital, Kobe, Hyogo 650-0047, Japan; [email protected] (A.I.); [email protected] (A.Y.); [email protected] (K.K.); [email protected] (Y.H.); [email protected] (Y.K.); [email protected] (M.T.) 2 Laboratory for Retinal Regeneration, RIKEN, Center for Biosystems Dynamics Research, Kobe, Hyogo 650-0047, Japan 3 Department of Medical Ethics, Graduate School of Medicine, Kyoto University, Kyoto 606-8501, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-(0)78-306-3305 Received: 11 September 2020; Accepted: 19 October 2020; Published: 22 October 2020 Abstract: USH2A is a common causal gene of retinitis pigmentosa (RP), a progressive blinding disease due to retinal degeneration. Genetic alterations in USH2A can lead to two types of RP, non-syndromic and syndromic RP, which is called Usher syndrome, with impairments of vision and hearing. The complexity of the genotype–phenotype correlation in USH2A-associated RP (USH2A-RP) has been reported. Genetic and clinical characterization of USH2A-RP has not been performed in Japanese patients. In this study, genetic analyses were performed using targeted panel sequencing in 525 Japanese RP patients. -
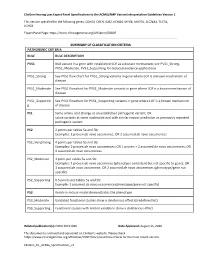
Clingen Hearing Loss Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1
ClinGen Hearing Loss Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1 This version specified for the following genes: CDH23, COCH, GJB2, KCNQ4, MYO6, MYO7A, SLC26A4, TECTA, USH2A Expert Panel Page: https://www.clinicalgenome.org/affiliation/50007 SUMMARY OF CLASSIFICATION CRITERIA PATHOGENIC CRITERIA RULE RULE DESCRIPTION PVS1 Null variant in a gene with established LOF as a disease mechanism; see PVS1_Strong, PVS1_Moderate, PVS1_Supporting for reduced evidence applications PVS1_Strong See PVS1 flow chart for PVS1_Strong variants in gene where LOF is a known mechanism of disease PVS1_Moderate See PVS1 flowchart for PVS1_Moderate variants in gene where LOF is a known mechanism of disease PVS1_Supportin See PVS1 flowchart for PVS1_Supporting variants in gene where LOF is a known mechanism g of disease PS1 Same amino acid change as an established pathogenic variant; OR splice variants at same nucleotide and with similar impact prediction as previously reported pathogenic variant PS2 2 points per tables 5a and 5b: Examples: 1 proven de novo occurrence; OR 2 assumed de novo occurrences PS2_VeryStrong 4 points per tables 5a and 5b: Examples: 2 proven de novo occurrences; OR 1 proven + 2 assumed de novo occurrences; OR 4 assumed de novo occurrences PS2_Moderate 1 point per tables 5a and 5b: Examples: 1 proven de novo occurrence (phenotype consistent but not specific to gene); OR 1 assumed de novo occurrence; OR 2 assumed de novo occurrences (phenotype/gene not specific) PS2_Supporting 0.5 points per tables 5a and 5b: Example: 1 assumed de novo occurrence (phenotype/gene not specific) PS3 Knock-in mouse model demonstrates the phenotype PS3_Moderate Validated functional studies show a deleterious effect (predefined list) PS3_Supporting Functional studies with limited validation show a deleterious effect Related publication(s): PMID 30311386 Date Approved: August 15, 2018 This document is archived and versioned on ClinGen’s website. -

Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the C
G C A T T A C G G C A T genes Article Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation 1, 2, 3,4 2 2,4, Andrej Zupan y , Ana Fakin y , Saba Battelino , Martina Jarc-Vidmar , Marko Hawlina * , Crystel Bonnet 5,6,7,8,9, Christine Petit 5,6,7,8,9,10 and Damjan Glavaˇc 1,* 1 Department of Molecular Genetics, Institute of Pathology, Faculty of Medicine, University of Ljubljana, 1000 Ljubljana, Slovenia; [email protected] 2 Eye Hospital, University Medical Centre Ljubljana, Grabloviˇceva46, 1000 Ljubljana, Slovenia; [email protected] (A.F.); [email protected] (M.J.-V.) 3 Department of Otorhinolaryngology and Cervicofacial Surgery, University Medical Centre Ljubljana, 1000 Ljubljana, Slovenia; [email protected] 4 Faculty of Medicine, University of Ljubljana, 1000 Ljubljana, Slovenia 5 Unité de Génétique et Physiologie de l’Audition, Institut Pasteur, 75015 Paris, France; [email protected] (C.B.); [email protected] (C.P.) 6 Unité Mixte de Recherche en Santé (UMRS) 1120, Institut National de la Santé et de la Recherche Médicale (INSERM), 75015 Paris, France 7 Complexité du Vivant, Sorbonne Universités, Université Pierre et Marie Curie, Université Paris 06, 75005 Paris, France 8 Institut de l’Audition, 75012 Paris, France 9 Syndrome de Usher et Autres Atteintes Rétino-Cochléaires, Institut de la Vision, 75012 Paris, France 10 Collège de France, 75005 Paris, France * Correspondence: [email protected] (M.H.); [email protected] (D.G.) The authors contributed equally to this work. -

Paternal Uniparental Heterodisomy with Partial Isodisomy Of
OPHTHALMIC MOLECULAR GENETICS SECTION EDITOR: EDWIN M. STONE, MD, PhD Paternal Uniparental Heterodisomy With Partial Isodisomy of Chromosome 1 in a Patient With Retinitis Pigmentosa Without Hearing Loss and a Missense Mutation in the Usher Syndrome Type II Gene USH2A Carlo Rivolta, PhD; Eliot L. Berson, MD; Thaddeus P. Dryja, MD Objective: To evaluate a form of nonmendelian inher- cue or gamete complementation. A paternal second cousin itance in a patient with retinitis pigmentosa (RP). of the patient also had RP and also had an identical het- erozygous mutation in the USH2A gene in the same codon. Methods: Direct DNA sequencing of the USH2A cod- However, the analysis of an isocoding polymorphism 20 ing region and microsatellite analysis of polymorphic base pairs away and closely linked microsatellite mark- markers from chromosome 1 and other chromosomes. ers in the patient and family members indicated that the 2 mutant alleles are unlikely to be identical by descent Results: A patient with RP without hearing loss caused and that the 2 relatives fortuitously had RP and a muta- by the homozygous mutation Cys759Phe in the USH2A tion in the same codon of the USH2A gene. gene on chromosome 1q was found to be the daughter of a noncarrier mother and a father who was heterozy- Conclusion: This family illustrates that recessive RP with- gous for this change. Further evaluation with microsat- out hearing loss can rarely be inherited from only 1 un- ellite markers revealed that the patient had inherited 2 affected carrier parent in a nonmendelian manner. copies of chromosome 1 from her father and none from her mother.