The Composition of the Perinatal Intestinal Microbiota in Cattle
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2018/064165 A2 (.Pdf)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/064165 A2 05 April 2018 (05.04.2018) W !P O PCT (51) International Patent Classification: Published: A61K 35/74 (20 15.0 1) C12N 1/21 (2006 .01) — without international search report and to be republished (21) International Application Number: upon receipt of that report (Rule 48.2(g)) PCT/US2017/053717 — with sequence listing part of description (Rule 5.2(a)) (22) International Filing Date: 27 September 2017 (27.09.2017) (25) Filing Language: English (26) Publication Langi English (30) Priority Data: 62/400,372 27 September 2016 (27.09.2016) US 62/508,885 19 May 2017 (19.05.2017) US 62/557,566 12 September 2017 (12.09.2017) US (71) Applicant: BOARD OF REGENTS, THE UNIVERSI¬ TY OF TEXAS SYSTEM [US/US]; 210 West 7th St., Austin, TX 78701 (US). (72) Inventors: WARGO, Jennifer; 1814 Bissonnet St., Hous ton, TX 77005 (US). GOPALAKRISHNAN, Vanch- eswaran; 7900 Cambridge, Apt. 10-lb, Houston, TX 77054 (US). (74) Agent: BYRD, Marshall, P.; Parker Highlander PLLC, 1120 S. Capital Of Texas Highway, Bldg. One, Suite 200, Austin, TX 78746 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Gut Microbiota Features Associated with Clostridioides Difficile Colonization in Dairy Calves
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Gut microbiota features associated with Clostridioides 2 difficile colonization in dairy calves 3 4 Laurel E. Redding1, Alexander S. Berry2,3, Nagaraju Indugu1, Elizabeth Huang1, Daniel P. Beiting3, Dipti 5 Pitta1 6 7 8 1Department of Clinical Sciences, University of Pennsylvania, School of Veterinary Medicine, Kennett 9 Square, PA, USA 10 11 2Division of Gastroenterology, Hepatology, and Nutrition, Children’s Hospital of Philadelphia, Philadelphia, 12 PA, USA 13 14 3Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, 15 USA 16 17 18 Corresponding author: Laurel Redding, [email protected] 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Diarrheal disease, a major cause of morbidity and mortality in dairy calves, is strongly associated with the 22 health and composition of the gut microbiome. Clostridioides difficile is an opportunistic pathogen that 23 proliferates and can produce enterotoxins when the host experiences gut dysbiosis. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Exploring the Diversity and Antimicrobial Potential of Marine Actinobacteria from the Comau Fjord in Northern Patagonia, Chile
ORIGINAL RESEARCH published: 19 July 2016 doi: 10.3389/fmicb.2016.01135 Exploring the Diversity and Antimicrobial Potential of Marine Actinobacteria from the Comau Fjord in Northern Patagonia, Chile Agustina Undabarrena 1, Fabrizio Beltrametti 2, Fernanda P. Claverías 1, Myriam González 1, Edward R. B. Moore 3, 4, Michael Seeger 1 and Beatriz Cámara 1* 1 Laboratorio de Microbiología Molecular y Biotecnología Ambiental, Departamento de Química & Centro de Biotecnología Daniel Alkalay Lowitt, Universidad Técnica Federico Santa María, Valparaíso, Chile, 2 Actygea S.r.l., Gerenzano, Italy, 3 Culture Collection University of Gothenburg (CCUG), Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden, 4 Department of Infectious Diseases, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden Edited by: Bioprospecting natural products in marine bacteria from fjord environments are attractive Learn-Han Lee, due to their unique geographical features. Although, Actinobacteria are well known Monash University Malaysia Campus, Malaysia for producing a myriad of bioactive compounds, investigations regarding fjord-derived Reviewed by: marine Actinobacteria are scarce. In this study, the diversity and biotechnological Atte Von Wright, potential of Actinobacteria isolated from marine sediments within the Comau University of Eastern Finland, Finland fjord, in Northern Chilean Patagonia, were assessed by culture-based approaches. Polpass Arul Jose, Central Salt and Marine Chemicals The 16S rRNA gene sequences revealed that members phylogenetically related Research Institute, India to the Micrococcaceae, Dermabacteraceae, Brevibacteriaceae, Corynebacteriaceae, *Correspondence: Microbacteriaceae, Dietziaceae, Nocardiaceae, and Streptomycetaceae families were Beatriz Cámara [email protected] present at the Comau fjord. A high diversity of cultivable Actinobacteria (10 genera) was retrieved by using only five different isolation media. -

Alterations in the Gut Bacterial Microbiome in Fungal Keratitis Patients
RESEARCH ARTICLE Alterations in the gut bacterial microbiome in fungal Keratitis patients Sama Kalyana Chakravarthy1☯, Rajagopalaboopathi Jayasudha1☯, Konduri Ranjith1, Anirban Dutta2, Nishal Kumar Pinna2, Sharmila S. Mande2, Savitri Sharma1, Prashant Garg3, Somasheila I. Murthy3, Sisinthy Shivaji1* 1 Jhaveri Microbiology Centre, Brien Holden Eye Research Centre, L. V. Prasad Eye Institute, Kallam Anji Reddy campus, Hyderabad, India, 2 Bio-Sciences R&D Division, TCS Research, Tata Consultancy Services Ltd., Pune, India, 3 Tej Kohli Cornea Institute, L. V. Prasad Eye Institute, Kallam Anji Reddy campus, Hyderabad, India a1111111111 a1111111111 ☯ These authors contributed equally to this work. a1111111111 * [email protected] a1111111111 a1111111111 Abstract Dysbiosis in the gut microbiome has been implicated in several diseases including auto- immune diseases, inflammatory diseases, cancers and mental disorders. Keratitis is an OPEN ACCESS inflammatory disease of the eye significantly contributing to corneal blindness in the devel- Citation: Kalyana Chakravarthy S, Jayasudha R, oping world. It would be worthwhile to investigate the possibility of dysbiosis in the gut micro- Ranjith K, Dutta A, Pinna NK, Mande SS, et al. (2018) Alterations in the gut bacterial microbiome biome being associated with Keratitis. Here, we have analyzed fungal and bacterial in fungal Keratitis patients. PLoS ONE 13(6): populations in stool samples through high-throughput sequencing of the ITS2 region for e0199640. https://doi.org/10.1371/journal. fungi and V3-V4 region of 16S rRNA gene for bacteria in healthy controls (HC, n = 31) and pone.0199640 patients with fungal keratitis (FK, n = 32). Candida albicans (2 OTUs), Aspergillus (1 OTU) Editor: Suresh Yenugu, University of Hyderabad, and 3 other denovo-OTUs were enriched in FK samples and an unclassified denovo-OTU INDIA was enriched in HC samples. -

Mapping the Bacterial Ecology on the Phyllosphere Of
bioRxiv preprint doi: https://doi.org/10.1101/494799; this version posted December 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Mapping the bacterial ecology on the phyllosphere of grass hay and the 2 potential hazards of soaking fodder for horse gut health 3 Meriel JS Moore-Colyer1, Annette Longland2, Patricia Harris3, Susan Crosthwaite4 4 1 Royal Agricultural University, Cirencester Gloucestershire, UK GL7 6JS. 5 2Equine and Livestock Nutrition Services, Pantafallen Fach, Tregaron, Ceredigion, Wales. SY25 6NF 6 3Mars Horsecare UK LTD; Equine Studies Group, Waltham Centre for Pet Nutrition, Waltham-on-the Wolds, 7 Leicestershire, UK. LE14 4RT 8 4 Faculty of Biology, Medicine and Health, University of Manchester, UK M13 9PT. 9 10 Corresponding author: *[email protected] ORCID 0000-0002-9172-9862 11 12 Short title: The bacterial ecology on the phyllosphere of dry and post-soaked grass hay for 13 horses 14 bioRxiv preprint doi: https://doi.org/10.1101/494799; this version posted December 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 15 Abstract 16 Globally hay is the preferred forage for stabled horses. Variable nutritional and hygienic quality stimulates pre- 17 feeding soaking to reduce dust and nutrients to reduce respiratory and metabolic disorders in horses. -

Marine Rare Actinomycetes: a Promising Source of Structurally Diverse and Unique Novel Natural Products
Review Marine Rare Actinomycetes: A Promising Source of Structurally Diverse and Unique Novel Natural Products Ramesh Subramani 1 and Detmer Sipkema 2,* 1 School of Biological and Chemical Sciences, Faculty of Science, Technology & Environment, The University of the South Pacific, Laucala Campus, Private Mail Bag, Suva, Republic of Fiji; [email protected] 2 Laboratory of Microbiology, Wageningen University & Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands * Correspondence: [email protected]; Tel.: +31-317-483113 Received: 7 March 2019; Accepted: 23 April 2019; Published: 26 April 2019 Abstract: Rare actinomycetes are prolific in the marine environment; however, knowledge about their diversity, distribution and biochemistry is limited. Marine rare actinomycetes represent a rather untapped source of chemically diverse secondary metabolites and novel bioactive compounds. In this review, we aim to summarize the present knowledge on the isolation, diversity, distribution and natural product discovery of marine rare actinomycetes reported from mid-2013 to 2017. A total of 97 new species, representing 9 novel genera and belonging to 27 families of marine rare actinomycetes have been reported, with the highest numbers of novel isolates from the families Pseudonocardiaceae, Demequinaceae, Micromonosporaceae and Nocardioidaceae. Additionally, this study reviewed 167 new bioactive compounds produced by 58 different rare actinomycete species representing 24 genera. Most of the compounds produced by the marine rare actinomycetes present antibacterial, antifungal, antiparasitic, anticancer or antimalarial activities. The highest numbers of natural products were derived from the genera Nocardiopsis, Micromonospora, Salinispora and Pseudonocardia. Members of the genus Micromonospora were revealed to be the richest source of chemically diverse and unique bioactive natural products. -
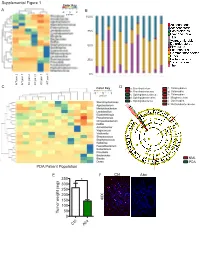
Mental Figure 1 Color Key a -2 0 2 B Z-Score 100%
Supplemental Figure 1 Color Key A -2 0 2 B z-score 100% 75% 50% 25% 0% KC pan 1 WT pan 3 WT KC pan 3 WT pan 2 WT pan 1 WT KC pan 2 C Color Key D a: Brevibacterium f: Chlamydiales b: Brevibacteriaceae g: Chlamydiia -3 0 3 z-score c: Sphingobacteriaceae h: Chlamydiae d: Sphingobacteriales i: Mogibacterium e: Sphingobacteriia j: Oscillospira k: Methylobacteriaceae NML Control Microb.-entrained MΦ PDA PDA Patient Population Control Microb.-entrained MΦ + Myd88i E F Ctrl Abx 350 * 300 250 200 150 40X 100 Tumor weight (mg) 50 0 x Ctrl Ab Supplemental Figure 2 A KC WT B ** * Actinobacteria * ** Bacteroidetes Cyanobacteria Deferribacteres * Firmicutes Proteobacteria % Relative abundance TM7 Others Time(wks) 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 Alpha Diversity Measure C E 60 KC WT 40 20 B. pseudolongum B. animalis 60 5 KC WT 0 B. adolescentis 40% Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 20 Age (weeks) B. pseudolongum B. animalis 5 0 B. adolescentis % Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 F Age (weeks) Week 3 Week 9 Week 13 p=0.678 p=0.02 p=0.385 Time(wks) 3 9 24 20 13 16 D 28 32 36 Week 13 KC WT Firmicutes; Ruminococcus Firmicutes; Dehalobacterium Alpha Diversity Measure Firmicutes; Oscillospira Bacteroidates; Odoribacter Axis.2 [12.7%] Actinobacteria; Bifidobacterium Axis.2 [23.8%] Axis.2 [24.7%] Week 16 Bacteroidetes; Bacteroidales Axis.1 [80.8%] Axis.1 [65.4%] Axis.1 [49.6%] Actinobacteria; Bifidobacterium Week 16 Week 20 Week 24 Week 20 p=0.339 p=0.036 p=0.021 Firmicutes; Dehalobacterium -

1 Supplementary Material a Major Clade of Prokaryotes with Ancient
Supplementary Material A major clade of prokaryotes with ancient adaptations to life on land Fabia U. Battistuzzi and S. Blair Hedges Data assembly and phylogenetic analyses Protein data set: Amino acid sequences of 25 protein-coding genes (“proteins”) were concatenated in an alignment of 18,586 amino acid sites and 283 species. These proteins included: 15 ribosomal proteins (RPL1, 2, 3, 5, 6, 11, 13, 16; RPS2, 3, 4, 5, 7, 9, 11), four genes (RNA polymerase alpha, beta, and gamma subunits, Transcription antitermination factor NusG) from the functional category of Transcription, three proteins (Elongation factor G, Elongation factor Tu, Translation initiation factor IF2) of the Translation, Ribosomal Structure and Biogenesis functional category, one protein (DNA polymerase III, beta subunit) of the DNA Replication, Recombination and repair category, one protein (Preprotein translocase SecY) of the Cell Motility and Secretion category, and one protein (O-sialoglycoprotein endopeptidase) of the Posttranslational Modification, Protein Turnover, Chaperones category, as annotated in the Cluster of Orthologous Groups (COG) (Tatusov et al. 2001). After removal of multiple strains of the same species, GBlocks 0.91b (Castresana 2000) was applied to each protein in the concatenation to delete poorly aligned sites (i.e., sites with gaps in more than 50% of the species and conserved in less than 50% of the species) with the following parameters: minimum number of sequences for a conserved position: 110, minimum number of sequences for a flank position: 110, maximum number of contiguous non-conserved positions: 32000, allowed gap positions: with half. The signal-to-noise ratio was determined by altering the “minimum length of a block” parameter. -

Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study Christopher J
OPEN LETTER https://doi.org/10.1038/s41586-018-0617-x Temporal development of the gut microbiome in early childhood from the TEDDY study Christopher J. Stewart1,2,18*, Nadim J. Ajami1,18, Jacqueline L. O’Brien1, Diane S. Hutchinson1, Daniel P. Smith1, Matthew C. Wong1, Matthew C. Ross1, Richard E. Lloyd1, HarshaVardhan Doddapaneni3, Ginger A. Metcalf3, Donna Muzny3, Richard A. Gibbs3, Tommi Vatanen4, Curtis Huttenhower4, Ramnik J. Xavier4, Marian Rewers5, William Hagopian6, Jorma Toppari7,8, Anette-G. Ziegler9,10,11, Jin-Xiong She12, Beena Akolkar13, Ake Lernmark14, Heikki Hyoty15,16, Kendra Vehik17, Jeffrey P. Krischer17 & Joseph F. Petrosino1* The development of the microbiome from infancy to childhood is sequencing, n = 10,867 samples from 783 children) and functional dependent on a range of factors, with microbial–immune crosstalk metagenome (metagenomic sequencing only) from longitudinal stool during this time thought to be involved in the pathobiology of later samples (Extended Data Table 1). A companion paper by Vatanen life diseases1–9 such as persistent islet autoimmunity and type 1 et al.14 focused exclusively on metagenomic sequencing data. diabetes10–12. However, to our knowledge, no studies have performed In this cohort of children that are at-risk for developing islet auto- extensive characterization of the microbiome in early life in a large, immunity (IA) or T1D, we aimed to (1) characterize definitively the multi-centre population. Here we analyse longitudinal stool samples longitudinal gut microbiome development from 3 to 46 months of age; from 903 children between 3 and 46 months of age by 16S rRNA gene (2) determine selected maternal and postnatal influences on the developing sequencing (n = 12,005) and metagenomic sequencing (n = 10,867), bacterial community during this same time period of early development; as part of the The Environmental Determinants of Diabetes in the and (3) use a nested case–control analysis to investigate the potential of the Young (TEDDY) study. -

Bibliography
Bibliography Abella, C.A., X.P. Cristina, A. Martinez, I. Pibernat and X. Vila. 1998. on moderate concentrations of acetate: production of single cells. Two new motile phototrophic consortia: "Chlorochromatium lunatum" Appl. Microbiol. Biotechnol. 35: 686-689. and "Pelochromatium selenoides". Arch. Microbiol. 169: 452-459. Ahring, B.K, P. Westermann and RA. Mah. 1991b. Hydrogen inhibition Abella, C.A and LJ. Garcia-Gil. 1992. Microbial ecology of planktonic of acetate metabolism and kinetics of hydrogen consumption by Me filamentous phototrophic bacteria in holomictic freshwater lakes. Hy thanosarcina thermophila TM-I. Arch. Microbiol. 157: 38-42. drobiologia 243-244: 79-86. Ainsworth, G.C. and P.H.A Sheath. 1962. Microbial Classification: Ap Acca, M., M. Bocchetta, E. Ceccarelli, R Creti, KO. Stetter and P. Cam pendix I. Symp. Soc. Gen. Microbiol. 12: 456-463. marano. 1994. Updating mass and composition of archaeal and bac Alam, M. and D. Oesterhelt. 1984. Morphology, function and isolation terial ribosomes. Archaeal-like features of ribosomes from the deep of halobacterial flagella. ]. Mol. Biol. 176: 459-476. branching bacterium Aquifex pyrophilus. Syst. Appl. Microbiol. 16: 629- Albertano, P. and L. Kovacik. 1994. Is the genus LeptolynglYya (Cyano 637. phyte) a homogeneous taxon? Arch. Hydrobiol. Suppl. 105: 37-51. Achenbach-Richter, L., R Gupta, KO. Stetter and C.R Woese. 1987. Were Aldrich, H.C., D.B. Beimborn and P. Schönheit. 1987. Creation of arti the original eubacteria thermophiles? Syst. Appl. Microbiol. 9: 34- factual internal membranes during fixation of Methanobacterium ther 39. moautotrophicum. Can.]. Microbiol. 33: 844-849. Adams, D.G., D. Ashworth and B. -

Comprehensive Relationships Between Gut Microbiome and Faecal Metabolome in Individuals with Type 2 Diabetes and Its Complications
Endocrine (2019) 66:526–537 https://doi.org/10.1007/s12020-019-02103-8 ORIGINAL ARTICLE Comprehensive relationships between gut microbiome and faecal metabolome in individuals with type 2 diabetes and its complications 1 2 1 3 1 1 Lijuan Zhao ● Hongxiang Lou ● Ying Peng ● Shihong Chen ● Yulong Zhang ● Xiaobo Li Received: 22 June 2019 / Accepted: 26 September 2019 / Published online: 7 October 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose As the treatment regimens such as metformin could confound the correlation between type 2 diabetes (T2D) and gut microbiome, we should revisit the relationship between gut microbiota and T2D patients who are not currently treated with metformin. Methods The study recruited 65 T2D patients: 49 with and 16 without diabetic complications, and 35 healthy controls. We sequenced the 16S rRNA V3-V4 region of gut microbiota and detected metabolites based on liquid chromatography mass spectrometry (LC/MS) and gas chromatography mass spectrometry (GC/MS) in faecal samples. 1234567890();,: 1234567890();,: Results The composition of both the gut microbiota and faecal metabolites changed significantly with T2D patients. The abundance of Proteobacteria and the ratio of Firmicutes/Bacteroidetes were higher in T2D patients than healthy subjects, and the short chain fatty acids (SCFAs), bile acids and lipids of T2D patients were significantly disordered. Moreover, the abundances of certain SCFA-producing bacteria (Lachnospiraceae and Ruminococcaceae etc.) were significantly increased in T2D patients, while the faecal SCFAs concentrations were significantly decreased. It’s suggested that the role of SCFA- producing bacteria was not simply to produce SCFAs. Then we identified 44 microbial modules to explore the correlations between the gut microbiota and metabolic traits.