Trna Fragments Show Intertwining with Mrnas of Specific Repeat
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pathway-Specific Dopaminergic Deficits in a Mouse Model of Angelman Syndrome
Pathway-specific dopaminergic deficits in a mouse model of Angelman syndrome Thorfinn T. Riday, … , Benjamin D. Philpot, C.J. Malanga J Clin Invest. 2012;122(12):4544-4554. https://doi.org/10.1172/JCI61888. Research Article Neuroscience Angelman syndrome (AS) is a neurodevelopmental disorder caused by maternal deletions or mutations of the ubiquitin ligase E3A (UBE3A) allele and characterized by minimal verbal communication, seizures, and disorders of voluntary movement. Previous studies have suggested that abnormal dopamine neurotransmission may underlie some of these deficits, but no effective treatment currently exists for the core features of AS. A clinical trial of levodopa (l-DOPA) in AS is ongoing, although the underlying rationale for this treatment strategy has not yet been thoroughly examined in preclinical models. We found that AS model mice lacking maternal Ube3a (Ube3am–/p+ mice) exhibit behavioral deficits that correlated with abnormal dopamine signaling. These deficits were not due to loss of dopaminergic neurons or impaired dopamine synthesis. Unexpectedly, Ube3am–/p+ mice exhibited increased dopamine release in the mesolimbic pathway while also exhibiting a decrease in dopamine release in the nigrostriatal pathway, as measured with fast-scan cyclic voltammetry. These findings demonstrate the complex effects of UBE3A loss on dopamine signaling in subcortical motor pathways that may inform ongoing clinical trials of l-DOPA therapy in patients with AS. Find the latest version: https://jci.me/61888/pdf Research article Pathway-specific dopaminergic deficits in a mouse model of Angelman syndrome Thorfinn T. Riday,1,2 Elyse C. Dankoski,1 Michael C. Krouse,3 Eric W. Fish,3 Paul L. -

The Effect of Temperature Adaptation on the Ubiquitin–Proteasome Pathway in Notothenioid Fishes Anne E
© 2017. Published by The Company of Biologists Ltd | Journal of Experimental Biology (2017) 220, 369-378 doi:10.1242/jeb.145946 RESEARCH ARTICLE The effect of temperature adaptation on the ubiquitin–proteasome pathway in notothenioid fishes Anne E. Todgham1,*, Timothy A. Crombie2 and Gretchen E. Hofmann3 ABSTRACT proliferation to compensate for the effects of low temperature on ’ There is an accumulating body of evidence suggesting that the sub- aerobic metabolism (Johnston, 1989; O Brien et al., 2003; zero Antarctic marine environment places physiological constraints Guderley, 2004). Recently, there has been an accumulating body – on protein homeostasis. Levels of ubiquitin (Ub)-conjugated proteins, of literature to suggest that protein homeostasis the maintenance of – 20S proteasome activity and mRNA expression of many proteins a functional protein pool has been highly impacted by evolution involved in both the Ub tagging of damaged proteins as well as the under these cold and stable conditions. different complexes of the 26S proteasome were measured to Maintaining protein homeostasis is a fundamental physiological examine whether there is thermal compensation of the Ub– process, reflecting a dynamic balance in synthetic and degradation proteasome pathway in Antarctic fishes to better understand the processes. There are numerous lines of evidence to suggest efficiency of the protein degradation machinery in polar species. Both temperature compensation of protein synthesis in Antarctic Antarctic (Trematomus bernacchii, Pagothenia borchgrevinki)and invertebrates (Whiteley et al., 1996; Marsh et al., 2001; Robertson non-Antarctic (Notothenia angustata, Bovichtus variegatus) et al., 2001; Fraser et al., 2002) and fish (Storch et al., 2005). In notothenioids were included in this study to investigate the zoarcid fishes, it has been demonstrated that Antarctic eelpouts mechanisms of cold adaptation of this pathway in polar species. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Transcriptional Regulation of RKIP in Prostate Cancer Progression
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Sciences Transcriptional Regulation of RKIP in Prostate Cancer Progression Submitted by: Sandra Marie Beach In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences Examination Committee Major Advisor: Kam Yeung, Ph.D. Academic William Maltese, Ph.D. Advisory Committee: Sonia Najjar, Ph.D. Han-Fei Ding, M.D., Ph.D. Manohar Ratnam, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: May 16, 2007 Transcriptional Regulation of RKIP in Prostate Cancer Progression Sandra Beach University of Toledo ACKNOWLDEGMENTS I thank my major advisor, Dr. Kam Yeung, for the opportunity to pursue my degree in his laboratory. I am also indebted to my advisory committee members past and present, Drs. Sonia Najjar, Han-Fei Ding, Manohar Ratnam, James Trempe, and Douglas Pittman for generously and judiciously guiding my studies and sharing reagents and equipment. I owe extended thanks to Dr. William Maltese as a committee member and chairman of my department for supporting my degree progress. The entire Department of Biochemistry and Cancer Biology has been most kind and helpful to me. Drs. Roy Collaco and Hong-Juan Cui have shared their excellent technical and practical advice with me throughout my studies. I thank members of the Yeung laboratory, Dr. Sungdae Park, Hui Hui Tang, Miranda Yeung for their support and collegiality. The data mining studies herein would not have been possible without the helpful advice of Dr. Robert Trumbly. I am also grateful for the exceptional assistance and shared microarray data of Dr. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Open Dogan Phdthesis Final.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science ELUCIDATING BIOLOGICAL FUNCTION OF GENOMIC DNA WITH ROBUST SIGNALS OF BIOCHEMICAL ACTIVITY: INTEGRATIVE GENOME-WIDE STUDIES OF ENHANCERS A Dissertation in Biochemistry, Microbiology and Molecular Biology by Nergiz Dogan © 2014 Nergiz Dogan Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2014 ii The dissertation of Nergiz Dogan was reviewed and approved* by the following: Ross C. Hardison T. Ming Chu Professor of Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee David S. Gilmour Professor of Molecular and Cell Biology Anton Nekrutenko Professor of Biochemistry and Molecular Biology Robert F. Paulson Professor of Veterinary and Biomedical Sciences Philip Reno Assistant Professor of Antropology Scott B. Selleck Professor and Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School iii ABSTRACT Genome-wide measurements of epigenetic features such as histone modifications, occupancy by transcription factors and coactivators provide the opportunity to understand more globally how genes are regulated. While much effort is being put into integrating the marks from various combinations of features, the contribution of each feature to accuracy of enhancer prediction is not known. We began with predictions of 4,915 candidate erythroid enhancers based on genomic occupancy by TAL1, a key hematopoietic transcription factor that is strongly associated with gene induction in erythroid cells. Seventy of these DNA segments occupied by TAL1 (TAL1 OSs) were tested by transient transfections of cultured hematopoietic cells, and 56% of these were active as enhancers. Sixty-six TAL1 OSs were evaluated in transgenic mouse embryos, and 65% of these were active enhancers in various tissues. -

Hematopoietic Differentiation
Hematopoietic Differentiation A number of lineages of mature cells are derived by a series of steps from a common stem cell precursor. Myeloid cells including neutrophils and monocytes are important for early defense against infection and because of the occurrence of myeloid leukemias. Myeloid differentiation and activation offers a favorable system for experimental investigation. Some Models of Gene Expression in Myeloid Cells 1. Compare resting neutrophils with neutrophils exposed for two hours to either. – A. Non-pathogenic E. coli. – B. Yersinia pestis D27 (plague bacillus). – C. Non-pathogenic Yersinia D28. 2. Induced differentiation of MPRO promyelocytic cells. NeutrophilsNeutrophils Major frontline defense cells against invading pathogens. – Constitute 70% of the total blood leukocytes. – Terminally differentiated professional phagocytic cells with an antibacterial arsenal. – Use both oxygen dependent and oxygen independent killing mechanisms. Play a central role in 2000X mag inflammation. Distribution of IL-8 transcripts on neutrophils (in situ hybridization) A combination of two Cy3 (red)- A labeled oligonucleotides complementary to IL-8 transcripts were used for in situ hybridization with neutrophils incubated in the absence (A) or presence (B) of ten E. coli K12 organisms per neutrophil. BB Methods of Analysis 1. mRNA analysis. – A. 3’ end restriction fragment gel display. – B. Affymetrix 60k or 42k oligonucleotide arrays. 2. Protein analysis. 2D gel electrophoresis using a variety of ph ranges for isoelectric focusing. MALDI-TOF identification of proteins. Representative Segments of Display Gels of cDNA Fragments: (Left) Neutrophils and Monocytes Exposed to E. coli for 2 H; (Right) Neutrophils Exposed to Various Bacteria for 2 H Ec: E. coli K12; Yp (KIM5): Y. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -
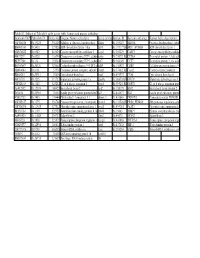
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1