Genomic Analysis of Helicobacter Himalayensis Sp. Nov. Isolated from Marmota Himalayana
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Food Or Beverage Product, Or Probiotic Composition, Comprising Lactobacillus Johnsonii 456
(19) TZZ¥¥¥ _T (11) EP 3 536 328 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 11.09.2019 Bulletin 2019/37 A61K 35/74 (2015.01) A61K 35/66 (2015.01) A61P 35/00 (2006.01) (21) Application number: 19165418.5 (22) Date of filing: 19.02.2014 (84) Designated Contracting States: • SCHIESTL, Robert, H. AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Encino, CA California 91436 (US) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • RELIENE, Ramune PL PT RO RS SE SI SK SM TR Los Angeles, CA California 90024 (US) • BORNEMAN, James (30) Priority: 22.02.2013 US 201361956186 P Riverside, CA California 92506 (US) 26.11.2013 US 201361909242 P • PRESLEY, Laura, L. Santa Maria, CA California 93458 (US) (62) Document number(s) of the earlier application(s) in • BRAUN, Jonathan accordance with Art. 76 EPC: Tarzana, CA California 91356 (US) 14753847.4 / 2 958 575 (74) Representative: Müller-Boré & Partner (71) Applicant: The Regents of the University of Patentanwälte PartG mbB California Friedenheimer Brücke 21 Oakland, CA 94607 (US) 80639 München (DE) (72) Inventors: Remarks: • YAMAMOTO, Mitsuko, L. This application was filed on 27-03-2019 as a Alameda, CA California 94502 (US) divisional application to the application mentioned under INID code 62. (54) FOOD OR BEVERAGE PRODUCT, OR PROBIOTIC COMPOSITION, COMPRISING LACTOBACILLUS JOHNSONII 456 (57) The present invention relates to food products, beverage products and probiotic compositions comprising Lacto- bacillus johnsonii 456. EP 3 536 328 A1 Printed by Jouve, 75001 PARIS (FR) EP 3 536 328 A1 Description CROSS-REFERENCE TO RELATED APPLICATIONS 5 [0001] This application claims the benefit of U.S. -

Journal of Clinical Microbiology
JOURNAL OF CLINICAL MICROBIOLOGY Volume 45 September 2007 No. 9 MINIREVIEW 16S rRNA Gene Sequencing for Bacterial Identification in J. Michael Janda and Sharon L. 2761–2764 the Diagnostic Laboratory: Pluses, Perils, and Pitfalls Abbott BACTERIOLOGY Is the Volume of Blood Cultured Still a Significant Factor in Emilio Bouza, Dolores Sousa, 2765–2769 the Diagnosis of Bloodstream Infections? Marta Rodrı´guez-Cre´ixems, Juan Garcı´a Lechuz, and Patricia Mun˜oz Reclassification of Phenotypically Identified Staphylococcus Takashi Sasaki, Ken Kikuchi, 2770–2778 intermedius Strains Yoshikazu Tanaka, Namiko Takahashi, Shinichi Kamata, and Keiichi Hiramatsu Evaluation of Gen-Probe APTIMA-Based Neisseria Erik Munson, Vivian Boyd, Jolanta 2793–2797 gonorrhoeae and Chlamydia trachomatis Confirmatory Testing Czarnecka, Judy Griep, Brian Lund, in a Metropolitan Setting of High Disease Prevalence Nancy Schaal, and Jeanne E. Hryciuk Convenient Test Using a Combination of Chelating Agents Soo-Young Kim, Seong Geun Hong, 2798–2801 for Detection of Metallo--Lactamases in the Clinical Ellen S. Moland, and Kenneth S. Laboratory Thomson Molecular Characterization of Vancomycin-Resistant Bo Zheng, Haruyoshi Tomita, Yong 2813–2818 Enterococcus faecium Isolates from Mainland China Hong Xiao, Shan Wang, Yun Li, and Yasuyoshi Ike Bacteriology of Moderate-to-Severe Diabetic Foot Infections Diane M. Citron, Ellie J. C. 2819–2828 and In Vitro Activity of Antimicrobial Agents Goldstein, C. Vreni Merriam, Benjamin A. Lipsky, and Murray A. Abramson Outbreak of Pseudomonas -

Westchester County Restaurants
RESTAURANTS THAT ARE ENOUGH TO MAKE YOU SICK: AN ANALYSIS OF UNSANITARY CONDITIONS AT NEW YORK CITY AND WESTCHESTER COUNTY RESTAURANTS STATE SENATOR JEFF KLEIN RANKING MINORITY MEMBER CONSUMER PROTECTION COMMITTEE RESTAURANTS ENOUGH TO MAKE YOU SICK: AN ANALYSIS OF UNSANITARY CONDITIONS AT NEW YORK CITY AND WESTCHESTER COUNTY RESTAURANT S How Consumers Are At Risk There is little reason why eating a meal in a restaurant should be any more dangerous than eating a meal in your own home. The volume and variety of foods prepared in a restaurant kitchen makes sanitation more critical, but the basic rules of cleanliness, temperature control and pest control are universal. The major difference is that the restaurant patron cannot usually see the kitchen where his or her meal is prepared. These unseen risks are present and they can pose a serious threat to the public. Worse, they may go undetected and unaddressed for extended periods of time. Although inspections are the first step toward catching these problems before they become public health hazards, many problems linger long after they have been cited by inspectors because restaurants can continue to operate with ongoing violations, with no warning to consumers. Many of the violations we have looked at, and many of the most common violations, are easily correctable, and if eradicated, greatly reduce the probability of an outbreak of food borne illness. By simply ensuring that potentially hazardous foods are properly stored, that they do not come into contact with other ready-to-eat foods, and ensuring that employees wash their hand or change their gloves after handling such foods would almost eliminate the risks posed by these foods. -

Genomics of Helicobacter Species 91
Genomics of Helicobacter Species 91 6 Genomics of Helicobacter Species Zhongming Ge and David B. Schauer Summary Helicobacter pylori was the first bacterial species to have the genome of two independent strains completely sequenced. Infection with this pathogen, which may be the most frequent bacterial infec- tion of humanity, causes peptic ulcer disease and gastric cancer. Other Helicobacter species are emerging as causes of infection, inflammation, and cancer in the intestine, liver, and biliary tract, although the true prevalence of these enterohepatic Helicobacter species in humans is not yet known. The murine pathogen Helicobacter hepaticus was the first enterohepatic Helicobacter species to have its genome completely sequenced. Here, we consider functional genomics of the genus Helico- bacter, the comparative genomics of the genus Helicobacter, and the related genera Campylobacter and Wolinella. Key Words: Cytotoxin-associated gene; H-Proteobacteria; gastric cancer; genomic evolution; genomic island; hepatobiliary; peptic ulcer disease; type IV secretion system. 1. Introduction The genus Helicobacter belongs to the family Helicobacteriaceae, order Campylo- bacterales, and class H-Proteobacteria, which is also known as the H subdivision of the phylum Proteobacteria. The H-Proteobacteria comprise of a relatively small and recently recognized line of descent within this extremely large and phenotypically diverse phy- lum. Other genera that colonize and/or infect humans and animals include Campylobac- ter, Arcobacter, and Wolinella. These organisms are all microaerophilic, chemoorgano- trophic, nonsaccharolytic, spiral shaped or curved, and motile with a corkscrew-like motion by means of polar flagella. Increasingly, free living H-Proteobacteria are being recognized in a wide range of environmental niches, including seawater, marine sedi- ments, deep-sea hydrothermal vents, and even as symbionts of shrimp and tubeworms in these environments. -

Co-Infection Associated with Diarrhea in a Colony of <I>Scid
Laboratory Animal Science Vol 48, No 5 Copyright 1998 October 1998 by the American Association for Laboratory Animal Science Helicobacter bilis/Helicobacter rodentium Co-Infection Associated with Diarrhea in a Colony of scid Mice Nirah H. Shomer,* Charles A. Dangler, Robert P. Marini, and James G. Fox† Abstract _ An outbreak of diarrhea spanning 3 months occurred in a breeding colony of scid/Trp53 knockout mice. Approximately a third of the 150 mice were clinically affected, with signs ranging from mucoid or watery diarrhea to severe hemorrhagic diarrhea with mortality. Helicobacter bilis and the newly recognized urease-negative organ- ism H. rodentium were isolated from microaerobic culture of feces or cecal specimens from affected mice. Dual infection with H. bilis and H. rodentium were confirmed by culture and polymerase chain reaction (PCR) in several animals. Both Helicobacter species rapidly colonized immunocompetent sentinel mice exposed to bedding from cages containing affected mice, but the sentinel remained asymptomatic. Mice with diarrhea had multifocal to segmental proliferative typhlitis, colitis, and proctitis. Several affected mice had multifocal mucosal necrosis with a few focal ulcers in the cecum, colon, and rectum. Mice with diarrhea were treated with antibiotic food wafers (1.5 mg of amoxicillin, 0.69 mg of metronidazole, and 0.185 mg of bismuth/mouse per day) previously shown to eradi- cate H. hepaticus in immunocompetent mice. Antibiotic treatment resulted in resolution of diarrhea, but not eradication of H. bilis and H. rodentium; mice continued to have positive PCR results after a 2-week treatment regimen, and clinical signs of diarrhea returned in some mice when treatment was suspended. -

List of the Pathogens Intended to Be Controlled Under Section 18 B.E
(Unofficial Translation) NOTIFICATION OF THE MINISTRY OF PUBLIC HEALTH RE: LIST OF THE PATHOGENS INTENDED TO BE CONTROLLED UNDER SECTION 18 B.E. 2561 (2018) By virtue of the provision pursuant to Section 5 paragraph one, Section 6 (1) and Section 18 of Pathogens and Animal Toxins Act, B.E. 2558 (2015), the Minister of Public Health, with the advice of the Pathogens and Animal Toxins Committee, has therefore issued this notification as follows: Clause 1 This notification is called “Notification of the Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2561 (2018).” Clause 2 This Notification shall come into force as from the following date of its publication in the Government Gazette. Clause 3 The Notification of Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2560 (2017) shall be cancelled. Clause 4 Define the pathogens codes and such codes shall have the following sequences: (1) English alphabets that used for indicating the type of pathogens are as follows: B stands for Bacteria F stands for fungus V stands for Virus P stands for Parasites T stands for Biological substances that are not Prion R stands for Prion (2) Pathogen risk group (3) Number indicating the sequence of each type of pathogens Clause 5 Pathogens intended to be controlled under Section 18, shall proceed as follows: (1) In the case of being the pathogens that are utilized and subjected to other law, such law shall be complied. (2) Apart from (1), the law on pathogens and animal toxin shall be complied. -
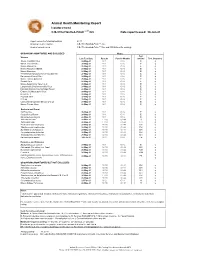
2021 ARC Health Report
Animal Health Monitoring Report Isolator reared C.B-17/IcrHanHsd-Prkdc scid /Arc Date report issued: 30-Jun-21 Report covers the following isolators: RI 27 Strains present in isolator: C.B-17/IcrHanHsd-Prkdc scid /Arc Strain of animal tested: C.B-17/IcrHanHsd-Prkdc scid /Arc and ICR Outbred for serology ORGANISMS MONITORED AND EXCLUDED Mouse Test Viruses Last Test Date Results Past 18 Months method Test frequency Mouse Hepatitis Virus 26-May-21 0 / 1 0 / 6 E q Minute Virus of Mice 26-May-21 0 / 1 0 / 6 E q Mouse Parvovirus 26-May-21 0 / 1 0 / 6 E q Murine Rotavirus (EDIM) 26-May-21 0 / 1 0 / 6 E q Mouse Norovirus 26-May-21 0 / 1 0 / 6 E q Theiler's Encephalomyelitis Virus (GD VII) 26-May-21 0 / 1 0 / 6 E q Pneumonia Virus of Mice 26-May-21 0 / 1 0 / 6 E q Murine Cytomegalovirus 26-May-21 0 / 1 0 / 6 E q Sendai Virus 26-May-21 0 / 1 0 / 6 E q Mouse Adenovirus Type 1 & 2 26-May-21 0 / 1 0 / 6 E q Lymphocytic Choriomeningitis Virus 26-May-21 0 / 1 0 / 6 E q Hantaan (Korean Haemorrhagic Fever) 26-May-21 0 / 1 0 / 6 E q Ectromelia (Mousepox) Virus 26-May-21 0 / 1 0 / 6 E q Reovirus -3 26-May-21 0 / 1 0 / 6 E q Polyoma Virus 26-May-21 0 / 1 0 / 6 E q K Virus 26-May-21 0 / 1 0 / 6 E q Lactic Dehydrogenase Elevating Virus 26-May-21 0 / 1 0 / 6 E q Mouse Thymic Virus 26-May-21 0 / 1 0 / 6 E q 00-Jan-00 Bacteria and Fungi CAR bacillus 26-May-21 0 / 1 0 / 6 E q Clostridium piliforme 26-May-21 0 / 1 0 / 6 E q Mycoplasma pulmonis 26-May-21 0 / 1 0 / 6 E q Helicobacter spp.1 26-May-21 0 / 10 0 / 44 H q Salmonella spp. -
R Graphics Output
883 | Desulfovibrio vulgaris | DvMF_2825 298701 | Desulfovibrio | DA2_3337 1121434 | Halodesulfovibrio aestuarii | AULY01000007_gene1045 207559 | Desulfovibrio alaskensis | Dde_0991 935942 | Desulfonatronum lacustre | KI912608_gene2193 159290 | Desulfonatronum | JPIK01000018_gene1259 1121448 | Desulfovibrio gigas | DGI_0655 1121445 | Desulfovibrio desulfuricans | ATUZ01000018_gene2316 525146 | Desulfovibrio desulfuricans | Ddes_0159 665942 | Desulfovibrio | HMPREF1022_02168 457398 | Desulfovibrio | HMPREF0326_00453 363253 | Lawsonia intracellularis | LI0397 882 | Desulfovibrio vulgaris | DVU_0784 1121413 | Desulfonatronovibrio hydrogenovorans | JMKT01000008_gene1463 555779 | Desulfonatronospira thiodismutans | Dthio_PD0935 690850 | Desulfovibrio africanus | Desaf_1578 643562 | Pseudodesulfovibrio aespoeensis | Daes_3115 1322246 | Pseudodesulfovibrio piezophilus | BN4_12523 641491 | Desulfovibrio desulfuricans | DND132_2573 1121440 | Desulfovibrio aminophilus | AUMA01000002_gene2198 1121456 | Desulfovibrio longus | ATVA01000018_gene290 526222 | Desulfovibrio salexigens | Desal_3460 1121451 | Desulfovibrio hydrothermalis | DESAM_21057 1121447 | Desulfovibrio frigidus | JONL01000008_gene3531 1121441 | Desulfovibrio bastinii | AUCX01000006_gene918 1121439 | Desulfovibrio alkalitolerans | dsat_0220 941449 | Desulfovibrio | dsx2_0067 1307759 | Desulfovibrio | JOMJ01000003_gene2163 1121406 | Desulfocurvus vexinensis | JAEX01000012_gene687 1304872 | Desulfovibrio magneticus | JAGC01000003_gene2904 573370 | Desulfovibrio magneticus | DMR_04750 -

Helicobacter Cinaedi Induced Typhlocolitis in Rag-2-Deficient Mice
Helicobacter cinaedi Induced Typhlocolitis in Rag-2-Deficient Mice The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Shen, Zeli; Feng, Yan; Rickman, Barry and Fox, James G. “ Helicobacter Cinaedi Induced Typhlocolitis in Rag-2-Deficient Mice .” Helicobacter 20, no. 2 (November 2014): 146–155 © 2014 John Wiley & Sons Ltd As Published http://dx.doi.org/10.1111/hel.12179 Publisher Wiley Blackwell Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/109464 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ HHS Public Access Author manuscript Author Manuscript Author ManuscriptHelicobacter Author Manuscript. Author manuscript; Author Manuscript available in PMC 2016 April 01. Published in final edited form as: Helicobacter. 2015 April ; 20(2): 146–155. doi:10.1111/hel.12179. Helicobacter cinaedi Induced Typhlocolitis in Rag-2-Deficient Mice Zeli Shen, Yan Feng, Barry Rickman, and James G. Fox Division of Comparative Medicine, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Abstract Background—Helicobacter cinaedi, an enterohepatic helicobacter species (EHS), is an important human pathogen and is associated with a wide range of diseases, especially in immunocompromised patients. It has been convincingly demonstrated that innate immune response to certain pathogenic enteric bacteria is sufficient to initiate colitis and colon carcinogenesis in recombinase-activating gene (Rag)-2-deficient mice model. To better understand the mechanisms of human IBD and its association with development of colon cancer, we investigated whether H. cinaedi could induce pathological changes noted with murine enterohepatic helicobacter infections in the Rag2−/− mouse model. -

Novel Mechanisms and Therapies for Celiac Disease
NOVEL MECHANISMS AND THERAPIES FOR CELIAC DISEASE EXPLORING NOVEL MECHANISMS AND THERAPIES FOR CELIAC DISEASE By HEATHER J GALIPEAU, B.H.Sc A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by Heather J Galipeau, 2015 PhD Thesis- H.J. Galipeau; McMaster University- Medical Sciences DESCRIPTIVE NOTE Doctor of Philosophy (2015) McMaster University, Hamilton, Ontario (Medical Sciences) TITLE Exploring novel mechanisms and therapies for celiac disease AUTHOR Heather J. Galipeau, B.H.Sc SUPERVISOR Dr. Elena F. Verdu NUMBER OF PAGES xix, 314 ii PhD Thesis- H.J. Galipeau; McMaster University- Medical Sciences ABSTRACT The gastrointestinal tract forms the body’s largest interface with the external environment and is exposed to a vast amount of foreign material, including pathogenic and commensal bacteria, as well as food antigens. The gastrointestinal tract has multiple functions that are performed through complex interactions by its different components. It must be able to degrade food, absorb nutrients and eliminate waste, while at the same time maintain a balance between immune tolerance and protection against pathogenic and antigenic material. This concept of mucosally-induced tolerance is a key feature of the gut immune system, whereby a state of local and systemic unresponsiveness to food protein or systemic ignorance of commensal bacteria is maintained under homeostatic conditions through interactions between the host, dietary factors, and the intestinal microbiota. Dysfunctional interactions can lead to a breakdown in tolerance to otherwise innocuous antigens. One of the best characterized food sensitivities is celiac disease (CD). -

The Following Pages Constitute the Final, Accepted and Revised Manuscript of the Article
The following pages constitute the final, accepted and revised manuscript of the article: Nilsson, Hans-Olof and Pietroiusti, Antonio and Gabrielli, Maurizio and Zocco, Maria Assunta and Gasbarrini, Giovanni and Gasbarrini, Antonio “Helicobacter pylori and Extragastric Diseases - Other Helicobacters” Helicobacter. 2005;10 Suppl 1:54-65. Publisher: Blackwell Use of alternative location to go to the published version of the article requires journal subscription. Alternative location: http://dx.doi.org/10.1111/j.1523-5378.2005.00334.x HELICOBACTER PYLORI AND EXTRAGASTRIC DISEASES - OTHER HELICOBACTERS Hans-Olof Nilsson, Antonio Pietroiusti*, Maurizio Gabrielli#, Maria Assunta Zocco#, Giovanni Gasbarrini#, Antonio Gasbarrini# Department of Laboratory Medicine, Lund University, Lund, Sweden *Medical Semiology and Methodology, Department of Internal Medicine, Tor Vergata University, Rome, Italy #Department of Internal Medicine, Catholic University the Sacred Heart, Gemelli Hospital Rome, Italy Correspondence and reprints request to: Antonio Gasbarrini, MD Istituto di Patologia Speciale Medica Universita’ Cattolica del Sacro Cuore Policlinico Gemelli, Largo Gemelli 8, 00168 Rome, ITALY Telephone: 39-335-6873562 39-6-30154294 FAX: 39-6-35502775 E-mail: [email protected] 2 ABSTRACT The involvement of Helicobacter pylori in the pathogenesis of extragastric diseases continues to be an interesting topic in the field of Helicobacter-related pathology. Although conflicting findings have been reported for most of the disorders, a role of H. pylori seems to be important especially for the development of cardiovascular and hematologic disorders. Previously isolated human and animal Helicobacter sp. flexispira and ′Helicobacter heilmannii′ strains have been validated using polyphasic taxonomy. A novel enterohepatic helicobacter has been isolated from mastomys and mice, adding to the list of helicobacters that colonize the liver. -

Living Organisms Author Their Read-Write Genomes in Evolution
biology Review Living Organisms Author Their Read-Write Genomes in Evolution James A. Shapiro ID Department of Biochemistry and Molecular Biology, University of Chicago GCIS W123B, 979 E. 57th Street, Chicago, IL 60637, USA; [email protected]; Tel.: +1-773-702-1625 Academic Editor: Andrés Moya Received: 23 August 2017; Accepted: 28 November 2017; Published: 6 December 2017 Abstract: Evolutionary variations generating phenotypic adaptations and novel taxa resulted from complex cellular activities altering genome content and expression: (i) Symbiogenetic cell mergers producing the mitochondrion-bearing ancestor of eukaryotes and chloroplast-bearing ancestors of photosynthetic eukaryotes; (ii) interspecific hybridizations and genome doublings generating new species and adaptive radiations of higher plants and animals; and, (iii) interspecific horizontal DNA transfer encoding virtually all of the cellular functions between organisms and their viruses in all domains of life. Consequently, assuming that evolutionary processes occur in isolated genomes of individual species has become an unrealistic abstraction. Adaptive variations also involved natural genetic engineering of mobile DNA elements to rewire regulatory networks. In the most highly evolved organisms, biological complexity scales with “non-coding” DNA content more closely than with protein-coding capacity. Coincidentally, we have learned how so-called “non-coding” RNAs that are rich in repetitive mobile DNA sequences are key regulators of complex phenotypes. Both biotic and abiotic ecological challenges serve as triggers for episodes of elevated genome change. The intersections of cell activities, biosphere interactions, horizontal DNA transfers, and non-random Read-Write genome modifications by natural genetic engineering provide a rich molecular and biological foundation for understanding how ecological disruptions can stimulate productive, often abrupt, evolutionary transformations.