An Msh2 Point Mutation Uncouples DNA Mismatch Repair and Apoptosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA
Mechanisms in E. coli and Human Mismatch Repair Nobel Lecture, December 8, 2015 by Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA. he idea that mismatched base pairs occur in cells and that such lesions trig- T ger their own repair was suggested 50 years ago by Robin Holliday in the context of genetic recombination [1]. Breakage and rejoining of DNA helices was known to occur during this process [2], with precision of rejoining attributed to formation of a heteroduplex joint, a region of helix where the two strands are derived from the diferent recombining partners. Holliday pointed out that if this heteroduplex region should span a genetic diference between the two DNAs, then it will contain one or more mismatched base pairs. He invoked processing of such mismatches to explain the recombination-associated phenomenon of gene conversion [1], noting that “If there are enzymes which can repair points of damage in DNA, it would seem possible that the same enzymes could recognize the abnormality of base pairing, and by exchange reactions rectify this.” Direct evidence that mismatches provoke a repair reaction was provided by bacterial transformation experiments [3–5], and our interest in this efect was prompted by the Escherichia coli (E. coli) work done in Matt Meselson’s lab at Harvard. Using artifcially constructed heteroduplex DNAs containing multiple mismatched base pairs, Wagner and Meselson [6] demonstrated that mismatches elicit a repair reaction upon introduction into the E. coli cell. Tey also showed that closely spaced mismatches, mismatches separated by a 1000 base pairs or so, are usually repaired on the same DNA strand. -

Developmental Delay and the Methyl Binding Genes H Turner, F Macdonald, S Warburton, F Latif, T Webb
1of3 ELECTRONIC LETTER J Med Genet: first published as 10.1136/jmg.40.2.e13 on 1 February 2003. Downloaded from Developmental delay and the methyl binding genes H Turner, F MacDonald, S Warburton, F Latif, T Webb ............................................................................................................................. J Med Genet 2003;40:e13(http://www.jmedgenet.com/cgi/content/full/40/2/e13) he report by Amir et al1 that Rett syndrome (RS) is associ- children referred with a clinical diagnosis of Angelman ated with mutations in the MECP2 gene permitted labora- syndrome (AS) but without an abnormality in 15q11q13 tory diagnosis of this devastating yet common neurode- showed that 4/46 of the girls in one case8 and 5/40 in the T 9 velopmental disorder. Hitherto the paucity of familial cases of other actually had Rett syndrome. Of these nine probands, the syndrome and the failure to identify the syndrome in only one was later found to have a clinical presentation incon- males despite fairly wide clinical criteria had defined it as an sistent with the laboratory diagnosis. This may not be surpris- X linked dominant disorder with male lethality.2 Soon, ing given that in very small girls the two syndromes may however, reports from the few families in which RS is present with overlapping clinical features and so be difficult to segregating showed that male family members who inherited differentiate on clinical grounds alone. the same mutation in the MECP2 gene as their affected female Familial cases of developmental handicap -

DNA Repair with Its Consequences (E.G
Cell Science at a Glance 515 DNA repair with its consequences (e.g. tolerance and pathways each require a number of apoptosis) as well as direct correction of proteins. By contrast, O-alkylated bases, Oliver Fleck* and Olaf Nielsen* the damage by DNA repair mechanisms, such as O6-methylguanine can be Department of Genetics, Institute of Molecular which may require activation of repaired by the action of a single protein, Biology, University of Copenhagen, Øster checkpoint pathways. There are various O6-methylguanine-DNA Farimagsgade 2A, DK-1353 Copenhagen K, Denmark forms of DNA damage, such as base methyltransferase (MGMT). MGMT *Authors for correspondence (e-mail: modifications, strand breaks, crosslinks removes the alkyl group in a suicide fl[email protected]; [email protected]) and mismatches. There are also reaction by transfer to one of its cysteine numerous DNA repair pathways. Each residues. Photolyases are able to split Journal of Cell Science 117, 515-517 repair pathway is directed to specific Published by The Company of Biologists 2004 covalent bonds of pyrimidine dimers doi:10.1242/jcs.00952 types of damage, and a given type of produced by UV radiation. They bind to damage can be targeted by several a UV lesion in a light-independent Organisms are permanently exposed to pathways. Major DNA repair pathways process, but require light (350-450 nm) endogenous and exogenous agents that are mismatch repair (MMR), nucleotide as an energy source for repair. Another damage DNA. If not repaired, such excision repair (NER), base excision NER-independent pathway that can damage can result in mutations, diseases repair (BER), homologous recombi- remove UV-induced damage, UVER, is and cell death. -

Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy
Biomolecules 2015, 5, 2184-2193; doi:10.3390/biom5042184 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Review Arsenic Disruption of DNA Damage Responses—Potential Role in Carcinogenesis and Chemotherapy Clarisse S. Muenyi 1, Mats Ljungman 2 and J. Christopher States 3,* 1 Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY 40292, USA; E-Mail: [email protected] 2 Departments of Radiation Oncology and Environmental Health Sciences, University of Michigan, Ann Arbor, MI 48109-2800, USA; E-Mail: [email protected] 3 Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY 40292, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-502-852-5347; Fax: +1-502-852-3123. Academic Editors: Wolf-Dietrich Heyer, Thomas Helleday and Fumio Hanaoka Received: 14 August 2015 / Accepted: 9 September 2015 / Published: 24 September 2015 Abstract: Arsenic is a Class I human carcinogen and is widespread in the environment. Chronic arsenic exposure causes cancer in skin, lung and bladder, as well as in other organs. Paradoxically, arsenic also is a potent chemotherapeutic against acute promyelocytic leukemia and can potentiate the cytotoxic effects of DNA damaging chemotherapeutics, such as cisplatin, in vitro. Arsenic has long been implicated in DNA repair inhibition, cell cycle disruption, and ubiquitination dysregulation, all negatively impacting the DNA damage response and potentially contributing to both the carcinogenic and chemotherapeutic potential of arsenic. Recent studies have provided mechanistic insights into how arsenic interferes with these processes including disruption of zinc fingers and suppression of gene expression. -

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

Expression of MSH2 and MSH6 on a Tissue Microarray in Patients with Osteosarcoma
ANTICANCER RESEARCH 34: 6961-6972 (2014) Expression of MSH2 and MSH6 on a Tissue Microarray in Patients with Osteosarcoma THORSTEN JENTZSCH1,2, BERNHARD ROBL1, MAREN HUSMANN1, BEATA BODE-LESNIEWSKA3 and BRUNO FUCHS1 1Laboratory for Orthopedic Research, Department of Orthopedics, Balgrist University Hospital, Zürich, Switzerland; 2Division of Trauma Surgery, Department of Surgery, University Hospital Zürich, Zürich, Switzerland; 3Institute of Surgical Pathology, University Hospital Zürich, Zürich, Switzerland Abstract. Background/Aim: Reliable prognostic factors for and head (4, 5). Following neoadjuvant chemotherapy, surgical the outcome of patients with osteosarcoma (OS) remain elusive. tumor resections provide tissue specimens for the assessment We analyzed the relationship between immunohistochemical of tumor necrosis rate, histology, grading and evaluation of expression of deoxyribonucleic acid (DNA) mismatch repair tumor biomarkers before further adjuvant chemotherapy may (MMR) proteins, MutS protein homolog 2 (MSH2) and MSH6 be initiated (6-8). Despite advances in therapeutic approaches, using a tissue microarray (TMA) with respect to OS patient five-year overall survival rates have been reported as 78% (9) demographics and survival time. Materials and Methods: We and are usually lower for non-responders to chemotherapy retrieved tumor tissue specimens from bone tissue originating (10) and patients with metastasis (11, 12). from surgical primary tumor specimens of OS patients to Albeit the limited number of studies (8, 13-17) on tumor generate a TMA and stained sections with antibodies against biomarkers, these are important for treatment decisions, MSH2 and MSH6. Results: Tumor resections of 67 patients patient discrimination regarding the response to with a mean follow-up of 98 months were evaluated. MSH2 chemotherapy and the incidence of metastasis, prediction of was expressed in nine (13%), MSH6 in ten (15%) and patient survival times and patient education (18-21). -

Deficiency in DNA Mismatch Repair of Methylation Damage Is a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Deficiency in DNA mismatch repair of methylation damage is a major 2 mutational process in cancer 3 1 1 1 1 1,* 4 Hu Fang , Xiaoqiang Zhu , Jieun Oh , Jayne A. Barbour , Jason W. H. Wong 5 6 1School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of 7 Hong Kong, Hong Kong Special Administrative Region 8 9 *Correspondence: [email protected] 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Abstract 2 DNA mismatch repair (MMR) is essential for maintaining genome integrity with its 3 deficiency predisposing to cancer1. MMR is well known for its role in the post- 4 replicative repair of mismatched base pairs that escape proofreading by DNA 5 polymerases following cell division2. Yet, cancer genome sequencing has revealed that 6 MMR deficient cancers not only have high mutation burden but also harbour multiple 7 mutational signatures3, suggesting that MMR has pleotropic effects on DNA repair. The 8 mechanisms underlying these mutational signatures have remained unclear despite 9 studies using a range of in vitro4,5 and in vivo6 models of MMR deficiency. -

Clinical Significance of ERCC2, XPC, ERCC5 and XRCC3 Gene Polymorphisms in Diffuse Large B Cell Lymphoma
DOI: 10.14744/ejmi.2020.56831 EJMI 2020;4(3):332–340 Research Article Clinical Significance of ERCC2, XPC, ERCC5 and XRCC3 Gene Polymorphisms in Diffuse Large B Cell Lymphoma Aykut Bahceci,1 Semra Paydas,2 Melek Ergin,3 Gulsah Seydaoglu,4 Gulsum Ucar5 1Department of Medical Oncology, Dr. Ersin Arslan Training and Research Hospital, Gaziantep, Turkey 2Department of Medical Oncology, Cukurova University Faculty of Medicine, Adana, Turkey 3Department of Patology, Cukurova University Faculty of Medicine, Adana, Turkey 4Department of Biostatistics, Cukurova University Faculty of Medicine, Adana, Turkey 5Department of Pediatric Hematology, Cukurova University Faculty of Medicine, Adana, Turkey Abstract Objectives: DNA repair genes protects the genome from DNA damage both of endogenous and exogenous stress fac- tors. Due to DNA repair gene polymorphisms, there are differences in the repair capacity between several cancer types. The aim of this study is to evaluate the association between some of the DNA repair gene polymorphisms and clinical outcome in Diffuse Large B-Cell Lymphoma (DLBCL). Methods: The association between clinical factors including stage at diagnosis, extra-nodal involvement, tumor bur- den, bone marrow involvement, relapse status, disease-free/overall survival times and DNA repair gene polymorphisms including ERCC2 (Lys751Gln), XPC (Gln939Lys), ERCC5 (Asp1104His) and XRCC3 (Thr241Met) in 58 patients with DLBCL. T-Shift Real-Time PCR was used to detect these mutations. Results: The median survival times were 60 months and 109 months in patients with CC genotype and CA/AA geno- type of XPC gene polymorphism, respectively (p=0.017). More interestingly, median survival times were 9 months and 109 months in patients with CC (XPC)/CC (XRCC3) and CA/AA (XPC)/CT/TT (XRCC3) for both XPC and XRCC3 gene polymorphisms, respectively (p=0.004). -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -

DNA Excision Repair Proteins and Gadd45 As Molecular Players for Active DNA Demethylation
Cell Cycle ISSN: 1538-4101 (Print) 1551-4005 (Online) Journal homepage: http://www.tandfonline.com/loi/kccy20 DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation Dengke K. Ma, Junjie U. Guo, Guo-li Ming & Hongjun Song To cite this article: Dengke K. Ma, Junjie U. Guo, Guo-li Ming & Hongjun Song (2009) DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation, Cell Cycle, 8:10, 1526-1531, DOI: 10.4161/cc.8.10.8500 To link to this article: http://dx.doi.org/10.4161/cc.8.10.8500 Published online: 15 May 2009. Submit your article to this journal Article views: 135 View related articles Citing articles: 92 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=kccy20 Download by: [University of Pennsylvania] Date: 27 April 2017, At: 12:48 [Cell Cycle 8:10, 1526-1531; 15 May 2009]; ©2009 Landes Bioscience Perspective DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation Dengke K. Ma,1,2,* Junjie U. Guo,1,3 Guo-li Ming1-3 and Hongjun Song1-3 1Institute for Cell Engineering; 2Department of Neurology; and 3The Solomon Snyder Department of Neuroscience; Johns Hopkins University School of Medicine; Baltimore, MD USA Abbreviations: DNMT, DNA methyltransferases; PGCs, primordial germ cells; MBD, methyl-CpG binding protein; NER, nucleotide excision repair; BER, base excision repair; AP, apurinic/apyrimidinic; SAM, S-adenosyl methionine Key words: DNA demethylation, Gadd45, Gadd45a, Gadd45b, Gadd45g, 5-methylcytosine, deaminase, glycosylase, base excision repair, nucleotide excision repair DNA cytosine methylation represents an intrinsic modifica- silencing of gene activity or parasitic genetic elements (Fig. -
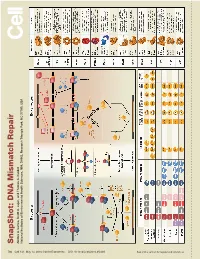
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases.