The Gut Microbiome of Exudivorous Wild and Captive Marmosets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded from Brill.Com09/27/2021 09:14:05PM Via Free Access 218 Rode-Margono & Nekaris – Impact of Climate and Moonlight on Javan Slow Lorises
Contributions to Zoology, 83 (4) 217-225 (2014) Impact of climate and moonlight on a venomous mammal, the Javan slow loris (Nycticebus javanicus Geoffroy, 1812) Eva Johanna Rode-Margono1, K. Anne-Isola Nekaris1, 2 1 Oxford Brookes University, Gipsy Lane, Headington, Oxford OX3 0BP, UK 2 E-mail: [email protected] Keywords: activity, environmental factors, humidity, lunarphobia, moon, predation, temperature Abstract Introduction Predation pressure, food availability, and activity may be af- To secure maintenance, survival and reproduction, fected by level of moonlight and climatic conditions. While many animals adapt their behaviour to various factors, such nocturnal mammals reduce activity at high lunar illumination to avoid predators (lunarphobia), most visually-oriented nocturnal as climate, availability of resources, competition, preda- primates and birds increase activity in bright nights (lunarphilia) tion, luminosity, habitat fragmentation, and anthropo- to improve foraging efficiency. Similarly, weather conditions may genic disturbance (Kappeler and Erkert, 2003; Beier influence activity level and foraging ability. We examined the 2006; Donati and Borgognini-Tarli, 2006). According response of Javan slow lorises (Nycticebus javanicus Geoffroy, to optimal foraging theory, animal behaviour can be seen 1812) to moonlight and temperature. We radio-tracked 12 animals as a trade-off between the risk of being preyed upon in West Java, Indonesia, over 1.5 years, resulting in over 600 hours direct observations. We collected behavioural and environmen- and the fitness gained from foraging (Charnov, 1976). tal data including lunar illumination, number of human observ- Perceived predation risk assessed through indirect cues ers, and climatic factors, and 185 camera trap nights on potential that correlate with the probability of encountering a predators. -

In Situ Conservation
NEWSN°17/DECEMBER 2020 Editorial IN SITU CONSERVATION One effect from 2020 is for sure: Uncertainty. Forward planning is largely News from the Little Fireface First, our annual SLOW event was impossible. We are acting and reacting Project, Java, Indonesia celebrated world-wide, including along the current situation caused by the By Prof K.A.I. Nekaris, MA, PhD by project partners Kukang Rescue Covid-19 pandemic. All zoos are struggling Director of the Little Fireface Project Program Sumatra, EAST Vietnam, Love economically after (and still ongoing) Wildlife Thailand, NE India Primate temporary closures and restricted business. The Little Fireface Project team has Investments in development are postponed Centre India, and the Bangladesh Slow at least. Each budget must be reviewed. been busy! Despite COVID we have Loris Project, to name a few. The end In the last newsletter we mentioned not been able to keep up with our wild of the week resulted in a loris virtual to forget about the support of the in situ radio collared slow lorises, including conference, featuring speakers from conservation efforts. Some of these under welcoming many new babies into the the helm of the Prosimian TAG are crucial 11 loris range countries. Over 200 for the survival of species – and for a more family. The ‘cover photo’ you see here people registered, and via Facebook sustainable life for the people involved in is Smol – the daughter of Lupak – and Live, more than 6000 people watched rd some of the poorest countries in the world. is our first 3 generation birth! Having the event. -
Online Appendix for “The Impact of the “World's 25 Most Endangered
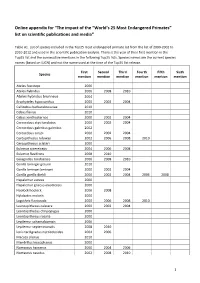
Online appendix for “The impact of the “World’s 25 Most Endangered Primates” list on scientific publications and media” Table A1. List of species included in the Top25 most endangered primate list from the list of 2000-2002 to 2010-2012 and used in the scientific publication analysis. There is the year of their first mention in the Top25 list and the consecutive mentions in the following Top25 lists. Species names are the current species names (based on IUCN) and not the name used at the time of the Top25 list release. First Second Third Fourth Fifth Sixth Species mention mention mention mention mention mention Ateles fusciceps 2006 Ateles hybridus 2006 2008 2010 Ateles hybridus brunneus 2004 Brachyteles hypoxanthus 2000 2002 2004 Callicebus barbarabrownae 2010 Cebus flavius 2010 Cebus xanthosternos 2000 2002 2004 Cercocebus atys lunulatus 2000 2002 2004 Cercocebus galeritus galeritus 2002 Cercocebus sanjei 2000 2002 2004 Cercopithecus roloway 2002 2006 2008 2010 Cercopithecus sclateri 2000 Eulemur cinereiceps 2004 2006 2008 Eulemur flavifrons 2008 2010 Galagoides rondoensis 2006 2008 2010 Gorilla beringei graueri 2010 Gorilla beringei beringei 2000 2002 2004 Gorilla gorilla diehli 2000 2002 2004 2006 2008 Hapalemur aureus 2000 Hapalemur griseus alaotrensis 2000 Hoolock hoolock 2006 2008 Hylobates moloch 2000 Lagothrix flavicauda 2000 2006 2008 2010 Leontopithecus caissara 2000 2002 2004 Leontopithecus chrysopygus 2000 Leontopithecus rosalia 2000 Lepilemur sahamalazensis 2006 Lepilemur septentrionalis 2008 2010 Loris tardigradus nycticeboides -

Science Journals
SCIENCE ADVANCES | REVIEW PRIMATOLOGY 2017 © The Authors, some rights reserved; Impending extinction crisis of the world’s primates: exclusive licensee American Association Why primates matter for the Advancement of Science. Distributed 1 2 3 4 under a Creative Alejandro Estrada, * Paul A. Garber, * Anthony B. Rylands, Christian Roos, Commons Attribution 5 6 7 7 Eduardo Fernandez-Duque, Anthony Di Fiore, K. Anne-Isola Nekaris, Vincent Nijman, NonCommercial 8 9 10 10 Eckhard W. Heymann, Joanna E. Lambert, Francesco Rovero, Claudia Barelli, License 4.0 (CC BY-NC). Joanna M. Setchell,11 Thomas R. Gillespie,12 Russell A. Mittermeier,3 Luis Verde Arregoitia,13 Miguel de Guinea,7 Sidney Gouveia,14 Ricardo Dobrovolski,15 Sam Shanee,16,17 Noga Shanee,16,17 Sarah A. Boyle,18 Agustin Fuentes,19 Katherine C. MacKinnon,20 Katherine R. Amato,21 Andreas L. S. Meyer,22 Serge Wich,23,24 Robert W. Sussman,25 Ruliang Pan,26 27 28 Inza Kone, Baoguo Li Downloaded from Nonhuman primates, our closest biological relatives, play important roles in the livelihoods, cultures, and religions of many societies and offer unique insights into human evolution, biology, behavior, and the threat of emerging diseases. They are an essential component of tropical biodiversity, contributing to forest regeneration and ecosystem health. Current information shows the existence of 504 species in 79 genera distributed in the Neotropics, mainland Africa, Madagascar, and Asia. Alarmingly, ~60% of primate species are now threatened with extinction and ~75% have de- clining populations. This situation is the result of escalating anthropogenic pressures on primates and their habitats— mainly global and local market demands, leading to extensive habitat loss through the expansion of industrial agri- http://advances.sciencemag.org/ culture, large-scale cattle ranching, logging, oil and gas drilling, mining, dam building, and the construction of new road networks in primate range regions. -

World's Most Endangered Primates
Primates in Peril The World’s 25 Most Endangered Primates 2016–2018 Edited by Christoph Schwitzer, Russell A. Mittermeier, Anthony B. Rylands, Federica Chiozza, Elizabeth A. Williamson, Elizabeth J. Macfie, Janette Wallis and Alison Cotton Illustrations by Stephen D. Nash IUCN SSC Primate Specialist Group (PSG) International Primatological Society (IPS) Conservation International (CI) Bristol Zoological Society (BZS) Published by: IUCN SSC Primate Specialist Group (PSG), International Primatological Society (IPS), Conservation International (CI), Bristol Zoological Society (BZS) Copyright: ©2017 Conservation International All rights reserved. No part of this report may be reproduced in any form or by any means without permission in writing from the publisher. Inquiries to the publisher should be directed to the following address: Russell A. Mittermeier, Chair, IUCN SSC Primate Specialist Group, Conservation International, 2011 Crystal Drive, Suite 500, Arlington, VA 22202, USA. Citation (report): Schwitzer, C., Mittermeier, R.A., Rylands, A.B., Chiozza, F., Williamson, E.A., Macfie, E.J., Wallis, J. and Cotton, A. (eds.). 2017. Primates in Peril: The World’s 25 Most Endangered Primates 2016–2018. IUCN SSC Primate Specialist Group (PSG), International Primatological Society (IPS), Conservation International (CI), and Bristol Zoological Society, Arlington, VA. 99 pp. Citation (species): Salmona, J., Patel, E.R., Chikhi, L. and Banks, M.A. 2017. Propithecus perrieri (Lavauden, 1931). In: C. Schwitzer, R.A. Mittermeier, A.B. Rylands, F. Chiozza, E.A. Williamson, E.J. Macfie, J. Wallis and A. Cotton (eds.), Primates in Peril: The World’s 25 Most Endangered Primates 2016–2018, pp. 40-43. IUCN SSC Primate Specialist Group (PSG), International Primatological Society (IPS), Conservation International (CI), and Bristol Zoological Society, Arlington, VA. -

Climate-Mediated Activity of the Javan Slow Loris, Nycticebus Javanicus
AIMS Environmental Science, 3(2): 249-260. DOI: 10.3934/environsci.2016.2.249 Received: 28 February 2016 Accepted: 21 April 2016 Published: 29 April 2016 http://www.aimspress.com/journal/environmental Research article Climate-mediated activity of the Javan Slow Loris, Nycticebus javanicus Kathleen D Reinhardt 1,3, Wirdateti 1,2,3 and K.AI. Nekaris 1, 3,* 1 Little Fireface Project; Cisurupan, Garut, Java, Indonesia 2 Research Centre for Biology-LIPI, Jakarta-Bogor, Cibinong, Indonesia 3 Nocturnal Primate Research Group, Oxford Brookes University, Oxford, UK * Corresponding: Email: [email protected]; Tel: +44 (0)-186-548-3767. Abstract: Joint impacts of anthropogenic disturbance and climate change are of pressing concern for modern conservationists. Climate change patterns have various diminishing effects on the biodiversity of an ecosystem, requiring an understanding of a species‘ ability to adapt. Agricultural practices are expanding at an altitudinal gradient on the Indonesian island of Java, forcing endemic species to range at increased elevation with lower temperatures, and in human-populated areas. One example is the Critically Endangered Javan slow loris (Nycticebus javanicus), which finds itself increasingly restricted to montane regions with extreme climate patterns and habitat disturbance. We observed wild N. javanicus in a highly fragmented, montane agroforest area to determine if climate variables and forest connectivity influence activity budget and behavior. Lorises ranged at different altitudes (1275 m above sea level (asl)—1570 m asl) and were observed for six months in Cipaganti, West Java. Using multinomial regression analyses, we found loris individuals were most likely to engage in increased foraging, feeding and travelling behavior than resting when relative humidity increases and in habitats with greater forest connectivity. -

The Effects of Habitat Disturbance on the Populations of Geoffroy's Spider Monkeys in the Yucatan Peninsula
The Effects of Habitat Disturbance on the Populations of Geoffroy’s Spider Monkeys in the Yucatan Peninsula PhD thesis Denise Spaan Supervisor: Filippo Aureli Co-supervisor: Gabriel Ramos-Fernández August 2017 Instituto de Neuroetología Universidad Veracruzana 1 For the spider monkeys of the Yucatan Peninsula, and all those dedicated to their conservation. 2 Acknowledgements This thesis turned into the biggest project I have ever attempted and it could not have been completed without the invaluable help and support of countless people and organizations. A huge thank you goes out to my supervisors Drs. Filippo Aureli and Gabriel Ramos- Fernández. Thank you for your guidance, friendship and encouragement, I have learnt so much and truly enjoyed this experience. This thesis would not have been possible without you and I am extremely proud of the results. Additionally, I would like to thank Filippo Aureli for all his help in organizing the logistics of field work. Your constant help and dedication to this project has been inspiring, and kept me pushing forward even when it was not always easy to do so, so thank you very much. I would like to thank Dr. Martha Bonilla for offering me an amazing estancia at the INECOL. Your kind words have encouraged and inspired me throughout the past three years, and have especially helped me to get through the last few months. Thank you! A big thank you to Drs. Colleen Schaffner and Jorge Morales Mavil for all your feedback and ideas over the past three years. Colleen, thank you for helping me to feel at home in Mexico and for all your support! I very much look forward to continue working with all of you in the future! I would like to thank the CONACYT for my PhD scholarship and the Instituto de Neuroetología for logistical, administrative and financial support. -

Fine Scale Habitat and Movement Patterns of Javan Slow Loris ( Nycticebus Javanicus )In Cipaganti, West Java, Indonesia
Fine scale habitat and movement patterns of javan slow loris ( Nycticebus javanicus )in Cipaganti, West Java, Indonesia Lina Fransson Degree project in biology, Master of science (2 years), 2018 Examensarbete i biologi 45 hp till masterexamen, 2018 Biology Education Centre, Uppsala University, and Department of Ecology and Genetics/ Animal Ecology, in cooperation with the Little Fireface Project Supervisors: Frank Johansson and Marie Sigaud External opponent: Josefine Stångberg Abstract....................................................................................................................................... 1 Introduction ................................................................................................................................ 2 Study questions ....................................................................................................................... 4 Materials and methods................................................................................................................ 5 Study area ............................................................................................................................... 5 “The species studied” in relation to its movement ................................................................. 6 Field work ............................................................................................................................... 8 Vegetation and habitat study ............................................................................................... -

Science Journals — AAAS
SCIENCE ADVANCES | REVIEW PRIMATOLOGY 2017 © The Authors, some rights reserved; Impending extinction crisis of the world’s primates: exclusive licensee American Association Why primates matter for the Advancement of Science. Distributed 1 2 3 4 under a Creative Alejandro Estrada, * Paul A. Garber, * Anthony B. Rylands, Christian Roos, Commons Attribution 5 6 7 7 Eduardo Fernandez-Duque, Anthony Di Fiore, K. Anne-Isola Nekaris, Vincent Nijman, NonCommercial 8 9 10 10 Eckhard W. Heymann, Joanna E. Lambert, Francesco Rovero, Claudia Barelli, License 4.0 (CC BY-NC). Joanna M. Setchell,11 Thomas R. Gillespie,12 Russell A. Mittermeier,3 Luis Verde Arregoitia,13 Miguel de Guinea,7 Sidney Gouveia,14 Ricardo Dobrovolski,15 Sam Shanee,16,17 Noga Shanee,16,17 Sarah A. Boyle,18 Agustin Fuentes,19 Katherine C. MacKinnon,20 Katherine R. Amato,21 Andreas L. S. Meyer,22 Serge Wich,23,24 Robert W. Sussman,25 Ruliang Pan,26 27 28 Inza Kone, Baoguo Li Downloaded from Nonhuman primates, our closest biological relatives, play important roles in the livelihoods, cultures, and religions of many societies and offer unique insights into human evolution, biology, behavior, and the threat of emerging diseases. They are an essential component of tropical biodiversity, contributing to forest regeneration and ecosystem health. Current information shows the existence of 504 species in 79 genera distributed in the Neotropics, mainland Africa, Madagascar, and Asia. Alarmingly, ~60% of primate species are now threatened with extinction and ~75% have de- clining populations. This situation is the result of escalating anthropogenic pressures on primates and their habitats— mainly global and local market demands, leading to extensive habitat loss through the expansion of industrial agri- http://advances.sciencemag.org/ culture, large-scale cattle ranching, logging, oil and gas drilling, mining, dam building, and the construction of new road networks in primate range regions. -

Central American Spider Monkey Ateles Geoffroyi Kuhl, 1820: Mexico, Guatemala, Nicaragua, Honduras, El Salvador
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/321428630 Central American Spider Monkey Ateles geoffroyi Kuhl, 1820: Mexico, Guatemala, Nicaragua, Honduras, El Salvador... Chapter · December 2017 CITATIONS READS 0 18 7 authors, including: Pedro Guillermo Mendez-Carvajal Gilberto Pozo-Montuy Fundacion Pro-Conservacion de los Primates… Conservación de la Biodiversidad del Usuma… 13 PUBLICATIONS 24 CITATIONS 32 PUBLICATIONS 202 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Connectivity of priority sites for primate conservation in the Zoque Rainforest Complex in Southeastern Mexico View project Regional Monitoring System: communitarian participation in surveying Mexican primate’s population (Ateles and Alouatta) View project All content following this page was uploaded by Gilberto Pozo-Montuy on 01 December 2017. The user has requested enhancement of the downloaded file. Primates in Peril The World’s 25 Most Endangered Primates 2016–2018 Edited by Christoph Schwitzer, Russell A. Mittermeier, Anthony B. Rylands, Federica Chiozza, Elizabeth A. Williamson, Elizabeth J. Macfie, Janette Wallis and Alison Cotton Illustrations by Stephen D. Nash IUCN SSC Primate Specialist Group (PSG) International Primatological Society (IPS) Conservation International (CI) Bristol Zoological Society (BZS) Published by: IUCN SSC Primate Specialist Group (PSG), International Primatological Society (IPS), Conservation International (CI), Bristol Zoological Society (BZS) Copyright: ©2017 Conservation International All rights reserved. No part of this report may be reproduced in any form or by any means without permission in writing from the publisher. Inquiries to the publisher should be directed to the following address: Russell A. -

Javan Slow Loris Nycticebus Javanicus) in a Montane Rainforest
Vol. 24: 95–103, 2014 ENDANGERED SPECIES RESEARCH Published online May 8 doi: 10.3354/esr00585 Endang Species Res Contribution to the Theme Section ‘Conservation and ecology of slow lorises’ FREEREE ACCESSCCESS Densities, distribution and detectability of a small nocturnal primate (Javan slow loris Nycticebus javanicus) in a montane rainforest K. Anne-Isola Nekaris1, Jarot Arisona Aji Pambudi2, Dwi Susanto2, Ridwan Djaffar Ahmad1,2, Vincent Nijman1,* 1Noctural Primate Research Group, Oxford Brookes University, OX3 0BP Oxford, UK 2Department of Biology, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok 16424, Indonesia ABSTRACT: Nocturnal mammals can be challenging to survey and, especially for many species that live in dense forest habitats, limited information is available on densities and distributions. We surveyed the endemic Javan slow loris Nycticebus javanicus in the montane forests of Mount Gede Pangrango, West Java, Indonesia. Surveys were conducted on 23 transects (260 h covering some 93 km) walking at variable speeds between 200 and 800 m h−1. Densities on individual tran- sects varied from 0 to 52 ind. km−2, with an overall density of 15.6 ind. km−2 (95% CI 9.7 to 25.2 ind. km−2). Encounter rates per kilometre were strongly influenced by the speed at which transects were walked, with fewer lorises detected at higher speeds. This effect was absent when consider- ing encounter rates per hour. Detectability and behavior of Javan slow lorises were not affected by the amount of lunar light and, in contrast to studies of some of their congeners, we found no evi- dence of lunar phobia or lunar philia. -

Blood Biochemistry Reference Values of Javan Slow Loris (Nycticebus Javanicus) in Rehabilitation Center
ACTA VETERINARIA INDONESIANA Special Issues: 127-131, May 2021 P-ISSN 2337-3202, E-ISSN 2337-4373 Research Blood Biochemistry Reference Values of Javan Slow Loris (Nycticebus javanicus) in Rehabilitation Center Nur Purba Priambada1*, Indri Saptorini1, Imam Arifin1, Wendi Prameswari1, Ursula Halla1, Karmele Llano Sanchez1, Joost Philippa1 1Yayasan Inisiasi Alam Rehabilitasi Indonesia, Jl. Curug Nangka, Sinarwangi RT 003/00, Kelurahan Sukajadi, Kecamatan Tamansari, Bogor, Jawa Barat, Indonesia - 16610 *Corresponding author: [email protected] Submitted April 15, 2021 Accepted May 16, 2021 ABSTRACT The Javan slow loris (Nycticebus javanicus) is an endemic primate species to Java Island, Indonesia. Currently, their conservation status is critically endangered due to habitat loss and the illegal wildlife trade. As a consequence of the pet trade, wild-caught slow lorises are confiscated or handed over to centers like IAR Indonesia Rescue Center. Rescued lorises present multiple health issues following stress, trauma, and miss- treatment after being kept as pets. During the medical evaluation, besides physical examination, blood biochemistry provides valuable diagnostic information. However, data on physiological values are unavailable and therefore interpretation of results is difficult. The objective of this study was to establish blood biochemistry reference values for wild, rehabilitant healthy adult Javan slow lorises in captivity. We anesthetized 20 individuals of Javan slow loris (10 males and 10 females) for pre-release check-up procedures. Blood samples were collected for blood biochemistry analysis on an in-house Vetscan VS2 (Zoetis), after which the results were statistically analyzed for mean and standard deviation. Results showed different values between the male and female group, however, they were not significant (p>0,05).