Composition Dependent Phase Separation Underlies Directional Flux
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modelling and Numerical Simulation of Phase Separation in Polymer Modified Bitumen by Phase- Field Method
http://www.diva-portal.org Postprint This is the accepted version of a paper published in Materials & design. This paper has been peer- reviewed but does not include the final publisher proof-corrections or journal pagination. Citation for the original published paper (version of record): Zhu, J., Lu, X., Balieu, R., Kringos, N. (2016) Modelling and numerical simulation of phase separation in polymer modified bitumen by phase- field method. Materials & design, 107: 322-332 http://dx.doi.org/10.1016/j.matdes.2016.06.041 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:kth:diva-188830 ACCEPTED MANUSCRIPT Modelling and numerical simulation of phase separation in polymer modified bitumen by phase-field method Jiqing Zhu a,*, Xiaohu Lu b, Romain Balieu a, Niki Kringos a a Department of Civil and Architectural Engineering, KTH Royal Institute of Technology, Brinellvägen 23, SE-100 44 Stockholm, Sweden b Nynas AB, SE-149 82 Nynäshamn, Sweden * Corresponding author. Email: [email protected] (J. Zhu) Abstract In this paper, a phase-field model with viscoelastic effects is developed for polymer modified bitumen (PMB) with the aim to describe and predict the PMB storage stability and phase separation behaviour. The viscoelastic effects due to dynamic asymmetry between bitumen and polymer are represented in the model by introducing a composition-dependent mobility coefficient. A double-well potential for PMB system is proposed on the basis of the Flory-Huggins free energy of mixing, with some simplifying assumptions made to take into account the complex chemical composition of bitumen. -

Understanding Variation in Partition Coefficient, Kd, Values: Volume II
United States Office of Air and Radiation EPA 402-R-99-004B Environmental Protection August 1999 Agency UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium August 1999 A Cooperative Effort By: Office of Radiation and Indoor Air Office of Solid Waste and Emergency Response U.S. Environmental Protection Agency Washington, DC 20460 Office of Environmental Restoration U.S. Department of Energy Washington, DC 20585 NOTICE The following two-volume report is intended solely as guidance to EPA and other environmental professionals. This document does not constitute rulemaking by the Agency, and cannot be relied on to create a substantive or procedural right enforceable by any party in litigation with the United States. EPA may take action that is at variance with the information, policies, and procedures in this document and may change them at any time without public notice. Reference herein to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government. ii FOREWORD Understanding the long-term behavior of contaminants in the subsurface is becoming increasingly more important as the nation addresses groundwater contamination. Groundwater contamination is a national concern as about 50 percent of the United States population receives its drinking water from groundwater. -

Phase Diagrams and Phase Separation
Phase Diagrams and Phase Separation Books MF Ashby and DA Jones, Engineering Materials Vol 2, Pergamon P Haasen, Physical Metallurgy, G Strobl, The Physics of Polymers, Springer Introduction Mixing two (or more) components together can lead to new properties: Metal alloys e.g. steel, bronze, brass…. Polymers e.g. rubber toughened systems. Can either get complete mixing on the atomic/molecular level, or phase separation. Phase Diagrams allow us to map out what happens under different conditions (specifically of concentration and temperature). Free Energy of Mixing Entropy of Mixing nA atoms of A nB atoms of B AM Donald 1 Phase Diagrams Total atoms N = nA + nB Then Smix = k ln W N! = k ln nA!nb! This can be rewritten in terms of concentrations of the two types of atoms: nA/N = cA nB/N = cB and using Stirling's approximation Smix = -Nk (cAln cA + cBln cB) / kN mix S AB0.5 This is a parabolic curve. There is always a positive entropy gain on mixing (note the logarithms are negative) – so that entropic considerations alone will lead to a homogeneous mixture. The infinite slope at cA=0 and 1 means that it is very hard to remove final few impurities from a mixture. AM Donald 2 Phase Diagrams This is the situation if no molecular interactions to lead to enthalpic contribution to the free energy (this corresponds to the athermal or ideal mixing case). Enthalpic Contribution Assume a coordination number Z. Within a mean field approximation there are 2 nAA bonds of A-A type = 1/2 NcAZcA = 1/2 NZcA nBB bonds of B-B type = 1/2 NcBZcB = 1/2 NZ(1- 2 cA) and nAB bonds of A-B type = NZcA(1-cA) where the factor 1/2 comes in to avoid double counting and cB = (1-cA). -

Phase Separation Phenomena in Solutions of Polysulfone in Mixtures of a Solvent and a Nonsolvent: Relationship with Membrane Formation*
Phase separation phenomena in solutions of polysulfone in mixtures of a solvent and a nonsolvent: relationship with membrane formation* J. G. Wijmans, J. Kant, M. H. V. Mulder and C. A. Smolders Department of Chemical Technology, Twente University of Technology, PO Box 277, 7500 AE Enschede, The Netherlands (Received 22 October 1984) The phase separation phenomena in ternary solutions of polysulfone (PSI) in mixtures of a solvent and a nonsolvent (N,N-dimethylacetamide (DMAc) and water, in most cases) are investigated. The liquid-liquid demixing gap is determined and it is shown that its location in the ternary phase diagram is mainly determined by the PSf-nonsolvent interaction parameter. The critical point in the PSf/DMAc/water system lies at a high polymer concentration of about 8~o by weight. Calorimetric measurements with very concentrated PSf/DMAc/water solutions (prepared through liquid-liquid demixing, polymer concentration of the polymer-rich phase up to 60%) showed no heat effects in the temperature range of -20°C to 50°C. It is suggested that gelation in PSf systems is completely amorphous. The results are incorporated into a discussion of the formation of polysulfone membranes. (Keywords: polysulfone; solutions; liquid-liquid demixing; crystallization; membrane structures; membrane formation) INTRODUCTION THEORY The field of membrane filtration covers a broad range of Membrane formation different separation techniques such as: hyperfiltration, In the phase inversion process a membrane is made by reverse osmosis, ultrafiltration, microfiltration, gas sepa- casting a polymer solution on a support and then bringing ration and pervaporation. Each process makes use of the solution to phase separation by means of solvent specific membranes which must be suited for the desired outflow and/or nonsolvent inflow. -

Henry's Law Constants and Micellar Partitioning of Volatile Organic
38 J. Chem. Eng. Data 2000, 45, 38-47 Henry’s Law Constants and Micellar Partitioning of Volatile Organic Compounds in Surfactant Solutions Leland M. Vane* and Eugene L. Giroux United States Environmental Protection Agency, National Risk Management Research Laboratory, 26 West Martin Luther King Drive, Cincinnati, Ohio 45268 Partitioning of volatile organic compounds (VOCs) into surfactant micelles affects the apparent vapor- liquid equilibrium of VOCs in surfactant solutions. This partitioning will complicate removal of VOCs from surfactant solutions by standard separation processes. Headspace experiments were performed to quantify the effect of four anionic surfactants and one nonionic surfactant on the Henry’s law constants of 1,1,1-trichloroethane, trichloroethylene, toluene, and tetrachloroethylene at temperatures ranging from 30 to 60 °C. Although the Henry’s law constant increased markedly with temperature for all solutions, the amount of VOC in micelles relative to that in the extramicellar region was comparatively insensitive to temperature. The effect of adding sodium chloride and isopropyl alcohol as cosolutes also was evaluated. Significant partitioning of VOCs into micelles was observed, with the micellar partitioning coefficient (tendency to partition from water into micelle) increasing according to the following series: trichloroethane < trichloroethylene < toluene < tetrachloroethylene. The addition of surfactant was capable of reversing the normal sequence observed in Henry’s law constants for these four VOCs. Introduction have long taken advantage of the high Hc values for these compounds. In the engineering field, the most common Vast quantities of organic solvents have been disposed method of dealing with chlorinated solvents dissolved in of in a manner which impacts human health and the groundwater is “pump & treat”swithdrawal of ground- environment. -

Partition Coefficients in Mixed Surfactant Systems
Partition coefficients in mixed surfactant systems Application of multicomponent surfactant solutions in separation processes Vom Promotionsausschuss der Technischen Universität Hamburg-Harburg zur Erlangung des akademischen Grades Doktor-Ingenieur genehmigte Dissertation von Tanja Mehling aus Lohr am Main 2013 Gutachter 1. Gutachterin: Prof. Dr.-Ing. Irina Smirnova 2. Gutachterin: Prof. Dr. Gabriele Sadowski Prüfungsausschussvorsitzender Prof. Dr. Raimund Horn Tag der mündlichen Prüfung 20. Dezember 2013 ISBN 978-3-86247-433-2 URN urn:nbn:de:gbv:830-tubdok-12592 Danksagung Diese Arbeit entstand im Rahmen meiner Tätigkeit als wissenschaftliche Mitarbeiterin am Institut für Thermische Verfahrenstechnik an der TU Hamburg-Harburg. Diese Zeit wird mir immer in guter Erinnerung bleiben. Deshalb möchte ich ganz besonders Frau Professor Dr. Irina Smirnova für die unermüdliche Unterstützung danken. Vielen Dank für das entgegengebrachte Vertrauen, die stets offene Tür, die gute Atmosphäre und die angenehme Zusammenarbeit in Erlangen und in Hamburg. Frau Professor Dr. Gabriele Sadowski danke ich für das Interesse an der Arbeit und die Begutachtung der Dissertation, Herrn Professor Horn für die freundliche Übernahme des Prüfungsvorsitzes. Weiterhin geht mein Dank an das Nestlé Research Center, Lausanne, im Besonderen an Herrn Dr. Ulrich Bobe für die ausgezeichnete Zusammenarbeit und der Bereitstellung von LPC. Den Studenten, die im Rahmen ihrer Abschlussarbeit einen wertvollen Beitrag zu dieser Arbeit geleistet haben, möchte ich herzlichst danken. Für den außergewöhnlichen Einsatz und die angenehme Zusammenarbeit bedanke ich mich besonders bei Linda Kloß, Annette Zewuhn, Dierk Claus, Pierre Bräuer, Heike Mushardt, Zaineb Doggaz und Vanya Omaynikova. Für die freundliche Arbeitsatmosphäre, erfrischenden Kaffeepausen und hilfreichen Gespräche am Institut danke ich meinen Kollegen Carlos, Carsten, Christian, Mohammad, Krishan, Pavel, Raman, René und Sucre. -

Estimation of Distribution Coefficients from the Partition Coefficient and Pka Douglas C
Estimation of Distribution Coefficients from the Partition Coefficient and pKa Douglas C. Scott* and Jeffrey W. Clymer he use of partition coefficients has received much atten- tion in the assessment of relative lipophilicity and hy- drophilicity of a compound. Recent advances in com- putational chemistry have enabled scientists to estimate Tpartition coefficients for neutral species very easily. For ioniz- able species, the distribution coefficient is a more relevant pa- rameter; however, less effort has been applied to its assessment. Knowledge of the distribution coefficient and pKa is important The authors discuss for the basic characterization of a compound (1), particularly the relationship when assessing the compound’s potential to penetrate biolog- between the partition ical or lipid barriers (2). In addition, the majority of compounds coefficient and the are passively absorbed and a large number of new chemical en- distribution coefficient tities fail in the development stages because of related phar- and relate the two with the use of equations. macokinetic reasons (3). Assessing a compound’s relative lipophilicity dictates a formulation strategy that will ensure ab- To accurately explain this relationship, the sorption or tissue penetration, which ultimately will lead to de- authors consider the partitioning of both the livery of the drug to the site of action. In recognition of the ionized and un-ionized species.The presence value of knowing the extent of a drug’s gastrointestinal per- of both species in the oil phase necessitates a meation along with other necessary parameters, FDA has is- dissociative equilibrium among these species sued guidance for a waiver of in vivo bioavailability and bio- such as that in the water phase. -

Chapter 9: Other Topics in Phase Equilibria
Chapter 9: Other Topics in Phase Equilibria This chapter deals with relations that derive in cases of equilibrium between combinations of two co-existing phases other than vapour and liquid, i.e., liquid-liquid, solid-liquid, and solid- vapour. Each of these phase equilibria may be employed to overcome difficulties encountered in purification processes that exploit the difference in the volatilities of the components of a mixture, i.e., by vapour-liquid equilibria. As with the case of vapour-liquid equilibria, the objective is to derive relations that connect the compositions of the two co-existing phases as functions of temperature and pressure. 9.1 Liquid-liquid Equilibria (LLE) 9.1.1 LLE Phase Diagrams Unlike gases which are miscible in all proportions at low pressures, liquid solutions (binary or higher order) often display partial immiscibility at least over certain range of temperature, and composition. If one attempts to form a solution within that certain composition range the system splits spontaneously into two liquid phases each comprising a solution of different composition. Thus, in such situations the equilibrium state of the system is two phases of a fixed composition corresponding to a temperature. The compositions of two such phases, however, change with temperature. This typical phase behavior of such binary liquid-liquid systems is depicted in fig. 9.1a. The closed curve represents the region where the system exists Fig. 9.1 Phase diagrams for a binary liquid system showing partial immiscibility in two phases, while outside it the state is a homogenous single liquid phase. Take for example, the point P (or Q). -

1 Supplementary Material to the Paper “The Temperature and Pressure Dependence of Nickel Partitioning Between Olivine and Sili
SUPPLEMENTARY MATERIAL TO THE PAPER “THE TEMPERATURE AND PRESSURE DEPENDENCE OF NICKEL PARTITIONING BETWEEN OLIVINE AND SILICATE MELT” A. K. Matzen, M. B. Baker, J. R. Beckett, and E. M. Stolper. In this Supplement, we provide seven sections that expand and/or illustrate specific topics presented in the main text. The first section is a sample calculation using our preferred partitioning expression, equation (5), from the main text. Section 2 compares our fitted values of ∆ r HT ,P ∆ r ST ,P − ref ref and ref ref for equation (5) to those calculated using tabulated thermodynamic R R data. In Section 3, we present details of the mass-balance calculations used to evaluate the experiments from both this work and the literature, and in Section 4 we describe in detail the construction of the Filter-B dataset. Section 5 details the construction and fits of the regular solution partitioning models. Section 6 is a comparison of the results of this work and the Beattie-Jones family of models. Finally, Section 7 lists all of the references incorporated into our database on olivine-liquid Ni partitioning. 1. EXAMPLE CALCULATION USING OUR PREFERRED PARTITIONING EXPRESSION (EQUATION 5) FROM THE MAIN TEXT Although one of the simplest equations listed in the text, we think it useful to present a detailed accounting of how to use equation (5) along with the fitted parameters presented in Table 4 to ol /liq ol /liq ol liq predict DNi (where DNi = NiO /NiO , by wt.) given the temperature and compositions of the coexisting olivine and liquid. In our example calculation, we predict the partition coefficient for one of our 1.0 GPa experiments, Run 6. -
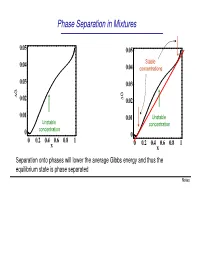
Phase Separation in Mixtures
Phase Separation in Mixtures 0.05 0.05 0.04 Stable 0.04 concentrations 0.03 0.03 G G ∆ 0.02 ∆ 0.02 0.01 0.01 Unstable Unstable concentration concentration 0 0 0 0.2 0.4 0.6 0.8 1 0 0.2 0.4 0.6 0.8 1 x x Separation onto phases will lower the average Gibbs energy and thus the equilibrium state is phase separated Notes Phase Separation versus Temperature Note that at higher temperatures the region of concentrations where phase separation takes place shrinks and G eventually disappears T increases ∆G = ∆U −T∆S This is because the term -T∆S becomes large at high temperatures. You can say that entropy “wins” over the potential energy cost at high temperatures Microscopically, the kinetic energy becomes x much larger than potential at high T, and the 1 x2 molecules randomly “run around” without noticing potential energy and thus intermix Oil and water mix at high temperature Notes Liquid-Gas Phase Separation in a Mixture A binary mixture can exist in liquid or gas phases. The liquid and gas phases have different Gibbs potentials as a function of mole fraction of one of the components of the mixture At high temperatures the Ggas <Gliquid because the entropy of gas is greater than that of liquid Tb2 T At lower temperatures, the two Gibbs b1 potentials intersect and separation onto gas and liquid takes place. Notes Boiling of a binary mixture Bubble point curve Dew point If we have a liquid at pint A that contains F A gas curve a molar fraction x1 of component 1 and T start heating it: E D - First the liquid heats to a temperature T’ C at constant x A and arrives to point B. -

Calculating the Partition Coefficients of Organic Solvents in Octanol/Water and Octanol/Air
Article Cite This: J. Chem. Inf. Model. XXXX, XXX, XXX−XXX pubs.acs.org/jcim Calculating the Partition Coefficients of Organic Solvents in Octanol/ Water and Octanol/Air † ‡ § ∥ Miroslava A. Nedyalkova,*, Sergio Madurga, Marek Tobiszewski, and Vasil Simeonov † Inorganic Chemistry Department, Faculty of Chemistry and Pharmacy, University of Sofia, Sofia 1164, Bulgaria ‡ Departament de Ciencià de Materials i Química Física and Institut de Química Teoricà i Computacional (IQTCUB), Universitat de Barcelona, 08028 Barcelona, Catalonia, Spain § Department of Analytical Chemistry, Faculty of Chemistry, Gdansḱ University of Technology (GUT), 80-233 Gdansk,́ Poland ∥ Analytical Chemistry Department, Faculty of Chemistry and Pharmacy, University of Sofia, Sofia 1164, Bulgaria *S Supporting Information ABSTRACT: Partition coefficients define how a solute is distributed between two immiscible phases at equilibrium. The experimental estimation of partition coefficients in a complex system can be an expensive, difficult, and time-consuming process. Here a computa- tional strategy to predict the distributions of a set of solutes in two relevant phase equilibria is presented. The octanol/water and octanol/air partition coefficients are predicted for a group of polar solvents using density functional theory (DFT) calculations in combination with a solvation model based on density (SMD) and are in excellent agreement with experimental data. Thus, the use of quantum-chemical calculations to predict partition coefficients from free energies should be a valuable alternative for unknown solvents. The obtained results indicate that the SMD continuum model in conjunction with any of the three DFT functionals (B3LYP, M06-2X, and M11) agrees with the observed experimental values. The highest correlation to experimental data for the octanol/water partition coefficients was reached by the M11 functional; for the octanol/air partition coefficient, the M06-2X functional yielded the best performance. -

Raoult's Law – Partition Law
BAE 820 Physical Principles of Environmental Systems Henry’s Law - Raoult's Law – Partition law Dr. Zifei Liu Biological and Agricultural Engineering Henry's law • At a constant temperature, the amount of a given gas that dissolves in a given type and volume of liquid is directly proportional to the partial pressure of that gas in equilibrium with that liquid. Pi = KHCi • Where Pi is the partial pressure of the gaseous solute above the solution, C is the i William Henry concentration of the dissolved gas and KH (1774-1836) is Henry’s constant with the dimensions of pressure divided by concentration. KH is different for each solute-solvent pair. Biological and Agricultural Engineering 2 Henry's law For a gas mixture, Henry's law helps to predict the amount of each gas which will go into solution. When a gas is in contact with the surface of a liquid, the amount of the gas which will go into solution is proportional to the partial pressure of that gas. An equivalent way of stating the law is that the solubility of a gas in a liquid is directly proportional to the partial pressure of the gas above the liquid. the solubility of gases generally decreases with increasing temperature. A simple rationale for Henry's law is that if the partial pressure of a gas is twice as high, then on the average twice as many molecules will hit the liquid surface in a given time interval, Biological and Agricultural Engineering 3 Air-water equilibrium Dissolution Pg or Cg Air (atm, Pa, mol/L, ppm, …) At equilibrium, Pg KH = Caq Water Caq (mol/L, mole ratio, ppm, …) Volatilization Biological and Agricultural Engineering 4 Various units of the Henry’s constant (gases in water at 25ºC) Form of K =P/C K =C /P K =P/x K =C /C equation H, pc aq H, cp aq H, px H, cc aq gas Units L∙atm/mol mol/(L∙atm) atm dimensionless -3 4 -2 O2 769 1.3×10 4.26×10 3.18×10 -4 4 -2 N2 1639 6.1×10 9.08×10 1.49×10 -2 3 CO2 29 3.4×10 1.63×10 0.832 Since all KH may be referred to as Henry's law constants, we must be quite careful to check the units, and note which version of the equation is being used.