An Improved Method for Utilizing High-Throughput Amplicon Sequencing to Determine the Diets of Insectivorous Animals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download The
AN ECOLOGICAL STUDY OF SOME OF THE CHIRONOMIDAE INHABITING A SERIES OF SALINE LAKES IN CENTRAL BRITISH COLUMBIA WITH SPECIAL REFERENCE TO CHIRONOMUS TENTANS FABRICIUS by Robert Alexander Cannings BSc. Hons., University of British Columbia, 1970 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in the Department of Zoology We accept this thesis as conforming to the required standard THE UNIVERSITY OF BRITISH COLUMBIA May, 1973 In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by his representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of The University of British Columbia Vancouver 8, Canada Date ii ABSTRACT This thesis is concerned with a study of the Chironomidae occuring in a saline lake series in central British Columbia. It describes the ecological distribution of species, their abundance, phenology and interaction, with particular attention being paid to Chironomus tentans. Emphasis is placed on the species of Chironomus that coexist in these lakes and a further analysis is made of the chromo• some inversion frequencies in C. tentans. Of the thirty-four species represented by identifiable adults in the study, eleven species have not been previously reported in British Columbia, five are new records for Canada and seven species are new to science. -

Water Ferns Azolla Spp. (Azollaceae) As New Host Plants for the Small China-Mark Moth, Cataclysta Lemnata (Linnaeus, 1758) (Lepidoptera, Crambidae, Acentropinae)
©Societas Europaea Lepidopterologica; download unter http://www.soceurlep.eu/ und www.zobodat.at Nota Lepi. 40(1) 2017: 1–13 | DOI 10.3897/nl.40.10062 Water ferns Azolla spp. (Azollaceae) as new host plants for the small China-mark moth, Cataclysta lemnata (Linnaeus, 1758) (Lepidoptera, Crambidae, Acentropinae) Atousa Farahpour-Haghani1,2, Mahdi Hassanpour1, Faramarz Alinia2, Gadir Nouri-Ganbalani1, Jabraeil Razmjou1, David Agassiz3 1 University of Mohaghegh Ardabili, Faculty of Agriculture and Natural Resources, Department of Plant Protection, Ardabil, Iran 2 Rice Research Institute of Iran (RRII), Agricultural Research, Education and Extension Organization (AREEO), Rasht, Iran 3 Department of Life Sciences, Natural History Museum, London SW7 5BD, England http://zoobank.org/307196B8-BB55-492B-8ECC-1F518D9EC9E4 Received 1 August 2016; accepted 3 November 2016; published: 20 January 2017 Subject Editor: Bernard Landry. Abstract. Water ferns (Azolla spp., Azollaceae) are reported for the first time as host plants for the larvae of the small China-mark moth Cataclysta lemnata (Linnaeus) (Lepidoptera: Crambidae: Acentropinae) in rice fields and waterways of northern Iran. Cataclysta lemnata is a semi-aquatic species that has been recorded to feed on Lemnaceae and a few other aquatic plants. However, it has not been reported before on Azolla spp. Larvae use water fern as food source and shelter and, at high population density in the laboratory, they completely wiped water fern from the water surface. Feeding was confirmed after rearing more than eight continual generations of C. lemnata on water fern in the laboratory. Adults obtained this way are darker and have darker fuscous markings in both sexes compared with specimens previously reported and the pattern remains unchanged after several generations. -

Water Mites of the Genus Arrenurus (Acari; Hydrachnida) from Europe and North America
Department of Animal Morphology Institute of Environmental Biology Adam Mickiewicz University Mariusz Więcek EFFECTS OF THE EVOLUTION OF INTROMISSION ON COURTSHIP COMPLEXITY AND MALE AND FEMALE MORPHOLOGY: WATER MITES OF THE GENUS ARRENURUS (ACARI; HYDRACHNIDA) FROM EUROPE AND NORTH AMERICA Mentors: Prof. Jacek Dabert – Institute of Environmental Biology, Adam Mickiewicz University Prof. Heather Proctor – Department of Biological Sciences, University of Alberta POZNAŃ 2015 1 ACKNOWLEDGEMENTS First and foremost I want to thank my mentor Prof. Jacek Dabert. It has been an honor to be his Ph.D. student. I would like to thank for his assistance and support. I appreciate the time and patience he invested in my research. My mentor, Prof. Heather Proctor, guided me into the field of behavioural biology, and advised on a number of issues during the project. She has been given me support and helped to carry through. I appreciate the time and effort she invested in my research. My research activities would not have happened without Prof. Lubomira Burchardt who allowed me to work in her team. Many thanks to Dr. Peter Martin who introduced me into the world of water mites. His enthusiasm was motivational and supportive, and inspirational discussions contributed to higher standard of my research work. I thank Dr. Mirosława Dabert for introducing me in to techniques of molecular biology. I appreciate Dr. Reinhard Gerecke and Dr. Harry Smit who provided research material for this study. Many thanks to Prof. Bruce Smith for assistance in identification of mites and sharing his expert knowledge in the field of pheromonal communication. I appreciate Dr. -

Bottom Fauna of the Mississippi River Near Keokuk, Iowa, with Particular Reference to Possible Control of Ephemeroptera and Tric
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1963 Bottom fauna of the Mississippi River near Keokuk, Iowa, with particular reference to possible control of Ephemeroptera and Trichoptera Clarence Albert Carlson Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Zoology Commons Recommended Citation Carlson, Clarence Albert, "Bottom fauna of the Mississippi River near Keokuk, Iowa, with particular reference to possible control of Ephemeroptera and Trichoptera " (1963). Retrospective Theses and Dissertations. 2956. https://lib.dr.iastate.edu/rtd/2956 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. BOTTOM FAUNA OF HIE MISSISSIPPI RIVER NEAR KEOKUK, IOWA, VttlH PARTICULAR REFERENCE TO POSSIBLE CONTROL OF EmEMEROPTBRA AND TRICHOPTBRA by Clarence Albert Carlson, Jr. A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF miLOSOHiY Major Subject: Zoology Approved: Signature was redacted for privacy. n Charge of Major Work Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Dean, College Iowa State University -

Lepidoptera of North America 5
Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera by Valerio Albu, 1411 E. Sweetbriar Drive Fresno, CA 93720 and Eric Metzler, 1241 Kildale Square North Columbus, OH 43229 April 30, 2004 Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Cover illustration: Blueberry Sphinx (Paonias astylus (Drury)], an eastern endemic. Photo by Valeriu Albu. ISBN 1084-8819 This publication and others in the series may be ordered from the C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences and Pest Management Colorado State University, Fort Collins, CO 80523 Abstract A list of 1531 species ofLepidoptera is presented, collected over 15 years (1988 to 2002), in eleven southern West Virginia counties. A variety of collecting methods was used, including netting, light attracting, light trapping and pheromone trapping. The specimens were identified by the currently available pictorial sources and determination keys. Many were also sent to specialists for confirmation or identification. The majority of the data was from Kanawha County, reflecting the area of more intensive sampling effort by the senior author. This imbalance of data between Kanawha County and other counties should even out with further sampling of the area. Key Words: Appalachian Mountains, -

Micro-Moth Grading Guidelines (Scotland) Abhnumber Code
Micro-moth Grading Guidelines (Scotland) Scottish Adult Mine Case ABHNumber Code Species Vernacular List Grade Grade Grade Comment 1.001 1 Micropterix tunbergella 1 1.002 2 Micropterix mansuetella Yes 1 1.003 3 Micropterix aureatella Yes 1 1.004 4 Micropterix aruncella Yes 2 1.005 5 Micropterix calthella Yes 2 2.001 6 Dyseriocrania subpurpurella Yes 2 A Confusion with fly mines 2.002 7 Paracrania chrysolepidella 3 A 2.003 8 Eriocrania unimaculella Yes 2 R Easier if larva present 2.004 9 Eriocrania sparrmannella Yes 2 A 2.005 10 Eriocrania salopiella Yes 2 R Easier if larva present 2.006 11 Eriocrania cicatricella Yes 4 R Easier if larva present 2.007 13 Eriocrania semipurpurella Yes 4 R Easier if larva present 2.008 12 Eriocrania sangii Yes 4 R Easier if larva present 4.001 118 Enteucha acetosae 0 A 4.002 116 Stigmella lapponica 0 L 4.003 117 Stigmella confusella 0 L 4.004 90 Stigmella tiliae 0 A 4.005 110 Stigmella betulicola 0 L 4.006 113 Stigmella sakhalinella 0 L 4.007 112 Stigmella luteella 0 L 4.008 114 Stigmella glutinosae 0 L Examination of larva essential 4.009 115 Stigmella alnetella 0 L Examination of larva essential 4.010 111 Stigmella microtheriella Yes 0 L 4.011 109 Stigmella prunetorum 0 L 4.012 102 Stigmella aceris 0 A 4.013 97 Stigmella malella Apple Pigmy 0 L 4.014 98 Stigmella catharticella 0 A 4.015 92 Stigmella anomalella Rose Leaf Miner 0 L 4.016 94 Stigmella spinosissimae 0 R 4.017 93 Stigmella centifoliella 0 R 4.018 80 Stigmella ulmivora 0 L Exit-hole must be shown or larval colour 4.019 95 Stigmella viscerella -

Data Quality, Performance, and Uncertainty in Taxonomic Identification for Biological Assessments
J. N. Am. Benthol. Soc., 2008, 27(4):906–919 Ó 2008 by The North American Benthological Society DOI: 10.1899/07-175.1 Published online: 28 October 2008 Data quality, performance, and uncertainty in taxonomic identification for biological assessments 1 2 James B. Stribling AND Kristen L. Pavlik Tetra Tech, Inc., 400 Red Brook Blvd., Suite 200, Owings Mills, Maryland 21117-5159 USA Susan M. Holdsworth3 Office of Wetlands, Oceans, and Watersheds, US Environmental Protection Agency, 1200 Pennsylvania Ave., NW, Mail Code 4503T, Washington, DC 20460 USA Erik W. Leppo4 Tetra Tech, Inc., 400 Red Brook Blvd., Suite 200, Owings Mills, Maryland 21117-5159 USA Abstract. Taxonomic identifications are central to biological assessment; thus, documenting and reporting uncertainty associated with identifications is critical. The presumption that comparable results would be obtained, regardless of which or how many taxonomists were used to identify samples, lies at the core of any assessment. As part of a national survey of streams, 741 benthic macroinvertebrate samples were collected throughout the eastern USA, subsampled in laboratories to ;500 organisms/sample, and sent to taxonomists for identification and enumeration. Primary identifications were done by 25 taxonomists in 8 laboratories. For each laboratory, ;10% of the samples were randomly selected for quality control (QC) reidentification and sent to an independent taxonomist in a separate laboratory (total n ¼ 74), and the 2 sets of results were compared directly. The results of the sample-based comparisons were summarized as % taxonomic disagreement (PTD) and % difference in enumeration (PDE). Across the set of QC samples, mean values of PTD and PDE were ;21 and 2.6%, respectively. -

Biological Monitoring of Surface Waters in New York State, 2019
NYSDEC SOP #208-19 Title: Stream Biomonitoring Rev: 1.2 Date: 03/29/19 Page 1 of 188 New York State Department of Environmental Conservation Division of Water Standard Operating Procedure: Biological Monitoring of Surface Waters in New York State March 2019 Note: Division of Water (DOW) SOP revisions from year 2016 forward will only capture the current year parties involved with drafting/revising/approving the SOP on the cover page. The dated signatures of those parties will be captured here as well. The historical log of all SOP updates and revisions (past & present) will immediately follow the cover page. NYSDEC SOP 208-19 Stream Biomonitoring Rev. 1.2 Date: 03/29/2019 Page 3 of 188 SOP #208 Update Log 1 Prepared/ Revision Revised by Approved by Number Date Summary of Changes DOW Staff Rose Ann Garry 7/25/2007 Alexander J. Smith Rose Ann Garry 11/25/2009 Alexander J. Smith Jason Fagel 1.0 3/29/2012 Alexander J. Smith Jason Fagel 2.0 4/18/2014 • Definition of a reference site clarified (Sect. 8.2.3) • WAVE results added as a factor Alexander J. Smith Jason Fagel 3.0 4/1/2016 in site selection (Sect. 8.2.2 & 8.2.6) • HMA details added (Sect. 8.10) • Nonsubstantive changes 2 • Disinfection procedures (Sect. 8) • Headwater (Sect. 9.4.1 & 10.2.7) assessment methods added • Benthic multiplate method added (Sect, 9.4.3) Brian Duffy Rose Ann Garry 1.0 5/01/2018 • Lake (Sect. 9.4.5 & Sect. 10.) assessment methods added • Detail on biological impairment sampling (Sect. -

Moths of North Carolina - Early Draft 1
Coleophoridae Blastobasis glandulella Acorn Moth 10 9 8 n=0 • 7 High Mt. • • 6 N 5 •• u 4 3 • • m 2 • • b 1 e 0 • • • r 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 NC counties: 15 Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec • • o 10 • f 9 n=4 = Sighting or Collection 8 • 7 Low Mt. High counts of: in NC since 2001 F 6 l 5 3 - Warren - 2015-05-12 4 i 3 1 - Ashe - 2012-08-20 g 2 Status Rank h 1 1 - Orange - 2015-06-09 0 NC US NC Global t 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 D Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec a 10 10 9 9 t 8 n=15 8 n=9 e 7 Pd 7 CP s 6 6 5 5 4 4 3 3 2 2 1 1 0 0 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 15 5 25 Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec Three periods to each month: 1-10 / 11-20 / 21-31 FAMILY: Coleophoridae SUBFAMILY: Blastobasinae TRIBE: Blastobasini TAXONOMIC_COMMENTS: FIELD GUIDE DESCRIPTIONS: Beadle and Leckie (2012); Leckie and Beadle (2018) ONLINE PHOTOS: TECHNICAL DESCRIPTION, ADULTS: Forbes (1923) TECHNICAL DESCRIPTION, IMMATURE STAGES: Galford (1986) ID COMMENTS: The following is based in part on the description by Forbes (1923). -
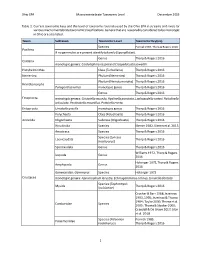
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

A Guide for the Identification of Two Subfamilies of Larval Chironomidae
Envlronment Canada Environnement Canada Fisheries Service des pêches .1 and Marine Service et des sciences de la mer L .' 1 '; ( 1 l r A Guide for the Identification of Two Subfamil ies of Larval Chironomidae: ,1"'--- The Chironominae and Tanypodlnae . : - - . ) / Found .in Benthic Studies Jin the / r~---.-_ c L___ r - - '" - .Ç"'''''-. Winnipeg River in the Vicinity ot Pine Falls, Manitoba in 1971 and 1972 by P. L. Stewart J.S. Loch Technical Report Series No. CEN/T-73-12 Resource Management Branch Central Region DEPARTMÈNT OF THE ENViRONMENT FISHERIES AND MARINE SERViCE Fisheries Operations Directorate Central Region Technical Reports Series No. CEN/T-73-12 A guide for the identification of two subfami lies of larva l Chironomidae~ the Chironominae and Tanypodinae found in benthic studies in the Winnipeg Riv~r in the vicinity of Pine Falls, Manitoba, in 1971 and 1972. by: P.L. Stewart qnd J.S. Loch ERRATA Page13: The caption for Figure 5A should read: Mentum and ventromental plates..•... instead of: submentum and ventromental plates..•.. Page 14: The caption for Figure 5B should read: Mentum and ventromental plates . instead of: submentum and ventromental plates.... DEPARTMENT OF THE ENVIRONMENT FISHERIES AND MARINE SERVICE Fisheries Operations Directorate Central Region Technical Report Series No: CEN/T-73-12 A GUIDE FOR THE IDENTIFICATION OF IWO SUBF.AMILIES OF LARVM.... CHIRONOMIDAE: THE CHIRONOMINAE AND TANYPODINAE FOUND IN BENTHIC STUDIES IN THE WINNIPEG RIVER IN THE vrCINITY OF PINE FM....LS, MANITOBA IN 1971 and 1972 by P. L. Stewart and J. S. Loch Resource Management Branch Fisheries Operations Directorate Central Region, Winnipeg November 1973 i ABSTRACT Identifying characteristics of the genera of two subfamilies of larvae of the midge family, C~onomldae (Vlpt~a), the C~ono mlnae and the Tanypodlnae, are presented with illustrations for the purpose of simplifying identification of these two groups by novice and more experienced personnel involved in assessment of benthic faunal composition. -

Recerca I Territori V12 B (002)(1).Pdf
Butterfly and moths in l’Empordà and their response to global change Recerca i territori Volume 12 NUMBER 12 / SEPTEMBER 2020 Edition Graphic design Càtedra d’Ecosistemes Litorals Mediterranis Mostra Comunicació Parc Natural del Montgrí, les Illes Medes i el Baix Ter Museu de la Mediterrània Printing Gràfiques Agustí Coordinadors of the volume Constantí Stefanescu, Tristan Lafranchis ISSN: 2013-5939 Dipòsit legal: GI 896-2020 “Recerca i Territori” Collection Coordinator Printed on recycled paper Cyclus print Xavier Quintana With the support of: Summary Foreword ......................................................................................................................................................................................................... 7 Xavier Quintana Butterflies of the Montgrí-Baix Ter region ................................................................................................................. 11 Tristan Lafranchis Moths of the Montgrí-Baix Ter region ............................................................................................................................31 Tristan Lafranchis The dispersion of Lepidoptera in the Montgrí-Baix Ter region ...........................................................51 Tristan Lafranchis Three decades of butterfly monitoring at El Cortalet ...................................................................................69 (Aiguamolls de l’Empordà Natural Park) Constantí Stefanescu Effects of abandonment and restoration in Mediterranean meadows .......................................87