Sapt2020: an Ab Initio Program for Symmetry-Adapted Perturbation Theory Calculations of Intermolecular Interaction Energies. Sequential and Parallel Versions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Density Functional Theory
Density Functional Approach Francesco Sottile Ecole Polytechnique, Palaiseau - France European Theoretical Spectroscopy Facility (ETSF) 22 October 2010 Density Functional Theory 1. Any observable of a quantum system can be obtained from the density of the system alone. < O >= O[n] Hohenberg, P. and W. Kohn, 1964, Phys. Rev. 136, B864 Density Functional Theory 1. Any observable of a quantum system can be obtained from the density of the system alone. < O >= O[n] 2. The density of an interacting-particles system can be calculated as the density of an auxiliary system of non-interacting particles. Hohenberg, P. and W. Kohn, 1964, Phys. Rev. 136, B864 Kohn, W. and L. Sham, 1965, Phys. Rev. 140, A1133 Density Functional ... Why ? Basic ideas of DFT Importance of the density Example: atom of Nitrogen (7 electron) 1. Any observable of a quantum Ψ(r1; ::; r7) 21 coordinates system can be obtained from 10 entries/coordinate ) 1021 entries the density of the system alone. 8 bytes/entry ) 8 · 1021 bytes 4:7 × 109 bytes/DVD ) 2 × 1012 DVDs 2. The density of an interacting-particles system can be calculated as the density of an auxiliary system of non-interacting particles. Density Functional ... Why ? Density Functional ... Why ? Density Functional ... Why ? Basic ideas of DFT Importance of the density Example: atom of Oxygen (8 electron) 1. Any (ground-state) observable Ψ(r1; ::; r8) 24 coordinates of a quantum system can be 24 obtained from the density of the 10 entries/coordinate ) 10 entries 8 bytes/entry ) 8 · 1024 bytes system alone. 5 · 109 bytes/DVD ) 1015 DVDs 2. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Popular GPU-Accelerated Applications
LIFE & MATERIALS SCIENCES GPU-ACCELERATED APPLICATIONS | CATALOG | AUG 12 LIFE & MATERIALS SCIENCES APPLICATIONS CATALOG Application Description Supported Features Expected Multi-GPU Release Status Speed Up* Support Bioinformatics BarraCUDA Sequence mapping software Alignment of short sequencing 6-10x Yes Available now reads Version 0.6.2 CUDASW++ Open source software for Smith-Waterman Parallel search of Smith- 10-50x Yes Available now protein database searches on GPUs Waterman database Version 2.0.8 CUSHAW Parallelized short read aligner Parallel, accurate long read 10x Yes Available now aligner - gapped alignments to Version 1.0.40 large genomes CATALOG GPU-BLAST Local search with fast k-tuple heuristic Protein alignment according to 3-4x Single Only Available now blastp, multi cpu threads Version 2.2.26 GPU-HMMER Parallelized local and global search with Parallel local and global search 60-100x Yes Available now profile Hidden Markov models of Hidden Markov Models Version 2.3.2 mCUDA-MEME Ultrafast scalable motif discovery algorithm Scalable motif discovery 4-10x Yes Available now based on MEME algorithm based on MEME Version 3.0.12 MUMmerGPU A high-throughput DNA sequence alignment Aligns multiple query sequences 3-10x Yes Available now LIFE & MATERIALS& LIFE SCIENCES APPLICATIONS program against reference sequence in Version 2 parallel SeqNFind A GPU Accelerated Sequence Analysis Toolset HW & SW for reference 400x Yes Available now assembly, blast, SW, HMM, de novo assembly UGENE Opensource Smith-Waterman for SSE/CUDA, Fast short -

Electronic Supporting Information a New Class of Soluble and Stable Transition-Metal-Substituted Polyoxoniobates: [Cr2(OH)4Nb10o
Electronic Supplementary Material (ESI) for Dalton Transactions This journal is © The Royal Society of Chemistry 2012 Electronic Supporting Information A New Class of Soluble and Stable Transition-Metal-Substituted Polyoxoniobates: [Cr2(OH)4Nb10O30]8-. Jung-Ho Son, C. André Ohlin and William H. Casey* A) Computational Results The two most interesting aspects of this new polyoxoanion is the inclusion of a more redox active metal and the optical activity in the visible region. For these reasons we in particular wanted to explore whether it was possible to readily and routinely predict the location of the absorbance bands of this type of compound. Also, owing to the difficulty in isolating the oxidised and reduced forms of the central polyanion dichromate species, we employed computations at the density functional theory (DFT) level to get a better idea of the potential characteristics of these forms. Structures were optimised for anti-parallel and parallel electronic spin alignments, and for both high and low spin cases where relevant. Intermediate spin configurations were 8- II III also tested for [Cr2(OH)4Nb10O30] . As expected, the high spin form of the Cr Cr complex was favoured (Table S1, below). DFT overestimates the bond lengths in general, but gives a good picture of the general distortion of the octahedral chromium unit, and in agreement with the crystal structure predicts that the shortest bond is the Cr-µ2-O bond trans from the central µ6- oxygen in the Lindqvist unit, and that the (Nb-)µ2-O-Cr-µ4-O(-Nb,Nb,Cr) angle is smaller by ca 5˚ from the ideal 180˚. -

Computer-Assisted Catalyst Development Via Automated Modelling of Conformationally Complex Molecules
www.nature.com/scientificreports OPEN Computer‑assisted catalyst development via automated modelling of conformationally complex molecules: application to diphosphinoamine ligands Sibo Lin1*, Jenna C. Fromer2, Yagnaseni Ghosh1, Brian Hanna1, Mohamed Elanany3 & Wei Xu4 Simulation of conformationally complicated molecules requires multiple levels of theory to obtain accurate thermodynamics, requiring signifcant researcher time to implement. We automate this workfow using all open‑source code (XTBDFT) and apply it toward a practical challenge: diphosphinoamine (PNP) ligands used for ethylene tetramerization catalysis may isomerize (with deleterious efects) to iminobisphosphines (PPNs), and a computational method to evaluate PNP ligand candidates would save signifcant experimental efort. We use XTBDFT to calculate the thermodynamic stability of a wide range of conformationally complex PNP ligands against isomeriation to PPN (ΔGPPN), and establish a strong correlation between ΔGPPN and catalyst performance. Finally, we apply our method to screen novel PNP candidates, saving signifcant time by ruling out candidates with non‑trivial synthetic routes and poor expected catalytic performance. Quantum mechanical methods with high energy accuracy, such as density functional theory (DFT), can opti- mize molecular input structures to a nearby local minimum, but calculating accurate reaction thermodynamics requires fnding global minimum energy structures1,2. For simple molecules, expert intuition can identify a few minima to focus study on, but an alternative approach must be considered for more complex molecules or to eventually fulfl the dream of autonomous catalyst design 3,4: the potential energy surface must be frst surveyed with a computationally efcient method; then minima from this survey must be refned using slower, more accurate methods; fnally, for molecules possessing low-frequency vibrational modes, those modes need to be treated appropriately to obtain accurate thermodynamic energies 5–7. -

Camcasp 5.9 Alston J. Misquitta† and Anthony J
CamCASP 5.9 Alston J. Misquittay and Anthony J. Stoneyy yDepartment of Physics and Astronomy, Queen Mary, University of London, 327 Mile End Road, London E1 4NS yy University Chemical Laboratory, Lensfield Road, Cambridge CB2 1EW September 1, 2015 Abstract CamCASP is a suite of programs designed to calculate molecular properties (multipoles and frequency- dependent polarizabilities) in single-site and distributed form, and interaction energies between pairs of molecules, and thence to construct atom–atom potentials. The CamCASP distribution also includes the programs Pfit, Casimir,Gdma 2.2, Cluster, and Process. Copyright c 2007–2014 Alston J. Misquitta and Anthony J. Stone Contents 1 Introduction 1 1.1 Authors . .1 1.2 Citations . .1 2 What’s new? 2 3 Outline of the capabilities of CamCASP and other programs 5 3.1 CamCASP limits . .7 4 Installation 7 4.1 Building CamCASP from source . .9 5 Using CamCASP 10 5.1 Workflows . 10 5.2 High-level scripts . 10 5.3 The runcamcasp.py script . 11 5.4 Low-level scripts . 13 6 Data conventions 13 7 CLUSTER: Detailed specification 14 7.1 Prologue . 15 7.2 Molecule definitions . 15 7.3 Geometry manipulations and other transformations . 16 7.4 Job specification . 18 7.5 Energy . 23 7.6 Crystal . 25 7.7 ORIENT ............................................... 26 7.8 Finally, . 29 8 Examples 29 8.1 A SAPT(DFT) calculation . 29 8.2 An example properties calculation . 30 8.3 Dispersion coefficients . 34 8.4 Using CLUSTER to obtain the dimer geometry . 36 9 CamCASP program specification 39 9.1 Global data . -

Dalton2018.0 – Lsdalton Program Manual
Dalton2018.0 – lsDalton Program Manual V. Bakken, A. Barnes, R. Bast, P. Baudin, S. Coriani, R. Di Remigio, K. Dundas, P. Ettenhuber, J. J. Eriksen, L. Frediani, D. Friese, B. Gao, T. Helgaker, S. Høst, I.-M. Høyvik, R. Izsák, B. Jansík, F. Jensen, P. Jørgensen, J. Kauczor, T. Kjærgaard, A. Krapp, K. Kristensen, C. Kumar, P. Merlot, C. Nygaard, J. Olsen, E. Rebolini, S. Reine, M. Ringholm, K. Ruud, V. Rybkin, P. Sałek, A. Teale, E. Tellgren, A. Thorvaldsen, L. Thøgersen, M. Watson, L. Wirz, and M. Ziolkowski Contents Preface iv I lsDalton Installation Guide 1 1 Installation of lsDalton 2 1.1 Installation instructions ............................. 2 1.2 Hardware/software supported .......................... 2 1.3 Source files .................................... 2 1.4 Installing the program using the Makefile ................... 3 1.5 The Makefile.config ................................ 5 1.5.1 Intel compiler ............................... 5 1.5.2 Gfortran/gcc compiler .......................... 9 1.5.3 Using 64-bit integers ........................... 12 1.5.4 Using Compressed-Sparse Row matrices ................ 13 II lsDalton User’s Guide 14 2 Getting started with lsDalton 15 2.1 The Molecule input file ............................ 16 2.1.1 BASIS ................................... 17 2.1.2 ATOMBASIS ............................... 18 2.1.3 User defined basis sets .......................... 19 2.2 The LSDALTON.INP file ............................ 21 3 Interfacing to lsDalton 24 3.1 The Grand Canonical Basis ........................... 24 3.2 Basisset and Ordering .............................. 24 3.3 Normalization ................................... 25 i CONTENTS ii 3.4 Matrix File Format ................................ 26 3.5 The lsDalton library bundle .......................... 27 III lsDalton Reference Manual 29 4 List of lsDalton keywords 30 4.1 **GENERAL ................................... 31 4.1.1 *TENSOR ............................... -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Quantum Chemistry on a Grid
Quantum Chemistry on a Grid Lucas Visscher Vrije Universiteit Amsterdam 1 Outline Why Quantum Chemistry needs to be gridified Computational Demands Other requirements What may be expected ? Program descriptions Dirac • Organization • Distributed development and testing Dalton Amsterdam Density Functional (ADF) An ADF-Dalton-Dirac grid driver Design criteria Current status Quantum Chemistry on the Grid, 08-11-2005 L. Visscher, Vrije Universiteit Amsterdam 2 Computational (Quantum) Chemistry QC is traditionally run on supercomputers Large fraction of the total supercomputer time annually allocated Characteristics Compute Time scales cubically or higher with number of atoms treated. Typical : a few hours to several days on a supercomputer Memory Usage scales quadratically or higher with number of atoms. Typical : A few hundred MB till tens of GB Data Storage variable. Typical: tens of Mb till hundreds of GB. Communication • Often serial (one-processor) runs in production work • Parallelization needs sufficient band width. Typical lower limit 100 Mb/s. Quantum Chemistry on the Grid, 08-11-2005 L. Visscher, Vrije Universiteit Amsterdam 3 Changing paradigms in quantum chemistry Computable molecules are often already “large” enough Many degrees of freedom Search for global minimum energy is less important Statistical sampling of different conformations is desired Let the nuclei move, follow molecules that evolve in time 2 steps : (Quantum) dynamics on PES 1 step : Classical dynamics with forces computed via QM Changing characteristics General: more similar calculations Compute time : Increases : more calculations Memory usage : Stays constant : typical size remains the same Data storage : Decreases : intermediate results are less important Communication: Decreases : only start-up data and final result Quantum Chemistry on the Grid, 08-11-2005 L. -
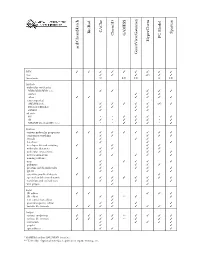
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -
![Computational Studies of the Unusual Water Adduct [Cp2time (OH2)]: the Roles of the Solvent and the Counterion](https://docslib.b-cdn.net/cover/1987/computational-studies-of-the-unusual-water-adduct-cp2time-oh2-the-roles-of-the-solvent-and-the-counterion-691987.webp)
Computational Studies of the Unusual Water Adduct [Cp2time (OH2)]: the Roles of the Solvent and the Counterion
Dalton Transactions View Article Online PAPER View Journal | View Issue Computational studies of the unusual water + adduct [Cp2TiMe(OH2)] : the roles of the Cite this: Dalton Trans., 2014, 43, 11195 solvent and the counterion† Jörg Saßmannshausen + The recently reported cationic titanocene complex [Cp2TiMe(OH2)] was subjected to detailed computational studies using density functional theory (DFT). The calculated NMR spectra revealed the importance of including the anion and the solvent (CD2Cl2) in order to calculate spectra which were in good agreement Received 29th January 2014, with the experimental data. Specifically, two organic solvent molecules were required to coordinate to Accepted 19th March 2014 the two hydrogens of the bound OH2 in order to achieve such agreement. Further elaboration of the role DOI: 10.1039/c4dt00310a of the solvent led to Bader’s QTAIM and natural bond order calculations. The zirconocene complex + www.rsc.org/dalton [Cp2ZrMe(OH2)] was simulated for comparison. Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Introduction Group 4 metallocenes have been the workhorse for a number of reactions for some decades now. In particular the cationic + compounds [Cp2MR] (M = Ti, Zr, Hf; R = Me, CH2Ph) are believed to be the active catalysts for a number of polymeriz- – ation reactions such as Kaminsky type α-olefin,1 10 11–15 16–18 This article is licensed under a carbocationic or ring-opening lactide polymerization. For these highly electrophilic cationic compounds, water is usually considered a poison as it leads to catalyst decompo- sition. In this respect it was quite surprising that Baird Open Access Article. Published on 21 March 2014.