Bioisosteric Replacement As a Tool in Anti-HIV Drug Design
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
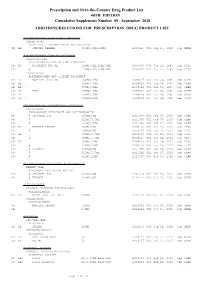
Additions and Deletions to the Drug Product List
Prescription and Over-the-Counter Drug Product List 40TH EDITION Cumulative Supplement Number 09 : September 2020 ADDITIONS/DELETIONS FOR PRESCRIPTION DRUG PRODUCT LIST ACETAMINOPHEN; BUTALBITAL; CAFFEINE TABLET;ORAL BUTALBITAL, ACETAMINOPHEN AND CAFFEINE >A> AA STRIDES PHARMA 325MG;50MG;40MG A 203647 001 Sep 21, 2020 Sep NEWA ACETAMINOPHEN; CODEINE PHOSPHATE SOLUTION;ORAL ACETAMINOPHEN AND CODEINE PHOSPHATE >D> AA WOCKHARDT BIO AG 120MG/5ML;12MG/5ML A 087006 001 Jul 22, 1981 Sep DISC >A> @ 120MG/5ML;12MG/5ML A 087006 001 Jul 22, 1981 Sep DISC TABLET;ORAL ACETAMINOPHEN AND CODEINE PHOSPHATE >A> AA NOSTRUM LABS INC 300MG;15MG A 088627 001 Mar 06, 1985 Sep CAHN >A> AA 300MG;30MG A 088628 001 Mar 06, 1985 Sep CAHN >A> AA ! 300MG;60MG A 088629 001 Mar 06, 1985 Sep CAHN >D> AA TEVA 300MG;15MG A 088627 001 Mar 06, 1985 Sep CAHN >D> AA 300MG;30MG A 088628 001 Mar 06, 1985 Sep CAHN >D> AA ! 300MG;60MG A 088629 001 Mar 06, 1985 Sep CAHN ACETAMINOPHEN; HYDROCODONE BITARTRATE TABLET;ORAL HYDROCODONE BITARTRATE AND ACETAMINOPHEN >A> @ CEROVENE INC 325MG;5MG A 211690 001 Feb 07, 2020 Sep CAHN >A> @ 325MG;7.5MG A 211690 002 Feb 07, 2020 Sep CAHN >A> @ 325MG;10MG A 211690 003 Feb 07, 2020 Sep CAHN >D> AA VINTAGE PHARMS 300MG;5MG A 090415 001 Jan 24, 2011 Sep DISC >A> @ 300MG;5MG A 090415 001 Jan 24, 2011 Sep DISC >D> AA 300MG;7.5MG A 090415 002 Jan 24, 2011 Sep DISC >A> @ 300MG;7.5MG A 090415 002 Jan 24, 2011 Sep DISC >D> AA 300MG;10MG A 090415 003 Jan 24, 2011 Sep DISC >A> @ 300MG;10MG A 090415 003 Jan 24, 2011 Sep DISC >D> @ XIROMED 325MG;5MG A 211690 -

Comparison of the Anti-Inflammatory Effects of Cilomilast, Budesonide
Ratcliffe and Dougall BMC Pharmacology and Toxicology 2012, 13:15 http://www.biomedcentral.com/2050-6511/13/15 RESEARCH ARTICLE Open Access Comparison of the anti-inflammatory effects of Cilomilast, Budesonide and a p38 Mitogen activated protein kinase inhibitor in COPD lung tissue macrophages Marianne Jennifer Ratcliffe1* and Iain Gordon Dougall2 Abstract Chronic Obstructive Pulmonary Disease (COPD) is a disease characterized by a largely irreversible airflow obstruction and a persistent, excessive inflammatory response. Alveolar macrophages (AMs) are increased in the lungs of COPD patients, and act as orchestrators of the inflammatory response, releasing a range of mediators to coordinate recruitment and activation of leukocytes. Attempts to treat the inflammatory component of COPD with anti-inflammatory drugs such as steroids has met with limited success. In this study, we compared the ability of the phosphodiesterase IV (PDEIV) inhibitor Cilomilast, the steroid Budesonide, and the p38 mitogen activated protein kinase inhibitor BIRB-796 to inhibit tumour necrosis factor alpha (TNFα) and interleukin 6 (IL-6) releases from AMs isolated from COPD lung transplant tissue. All studies were carried out with appropriate ethical approval and written, informed consent was obtained from each subject. Cilomilast had little effect on cytokine release from AMs. There was considerable variability in the responsiveness of AMs to Budesonide, with a subset of AMs responding poorly to Budesonide. BIRB-796 inhibited TNFα release from all AM donors, including those that responded poorly to steroids. Treatment with BIRB-796 and Budesonide together gave an additive decrease in TNFa release. These results suggest that a p38 inhibitor may provide advantages over existing anti-inflammatory treatments for COPD, either as an add-on to existing therapy, or to treat patients who respond poorly to steroids. -

Guanidine-Containing Polyhydroxyl Macrolides: Chemistry, Biology, and Structure-Activity Relationship
molecules Review Guanidine-Containing Polyhydroxyl Macrolides: Chemistry, Biology, and Structure-Activity Relationship Xiaoyuan Song 1, Ganjun Yuan 1,* , Peibo Li 2 and Sheng Cao 1 1 College of Bioscience and Bioengineering, Jiangxi Agricultural University, Nanchang 330045, China; [email protected] (X.S.); [email protected] (S.C.) 2 School of Life Sciences, Sun Yat-sen University, 135 Xingang Road, Guangzhou 510275, China; [email protected] * Correspondence: [email protected]; Tel.: +86-0791-83813459 Academic Editor: Jesus Simal-Gandara Received: 7 October 2019; Accepted: 29 October 2019; Published: 30 October 2019 Abstract: Antimicrobial resistance has been seriously threatening human health, and discovering new antimicrobial agents from the natural resource is still an important pathway among various strategies to prevent resistance. Guanidine-containing polyhydroxyl macrolides, containing a polyhydroxyl lactone ring and a guanidyl side chain, can be produced by many actinomycetes and have been proved to possess many bioactivities, especially broad-spectrum antibacterial and antifungal activities. To explore the potential of these compounds to be developed into new antimicrobial agents, a review on their structural diversities, spectroscopic characterizations, bioactivities, acute toxicities, antimicrobial mechanisms, and the structure-activity relationship was first performed based on the summaries and analyses of related publications from 1959 to 2019. A total of 63 guanidine-containing polyhydroxyl macrolides were reported, including -

Mediated Pulmonary Hypertension in Neonatal Rats: a Role for Products of Lipid Peroxidation
0031-3998/00/4803-0289 PEDIATRIC RESEARCH Vol. 48, No. 3, 2000 Copyright © 2000 International Pediatric Research Foundation, Inc. Printed in U.S.A. Endothelin-1 and O2-Mediated Pulmonary Hypertension in Neonatal Rats: A Role for Products of Lipid Peroxidation ROBERT P. JANKOV, XIAOPING LUO, JUDY CABACUNGAN, ROSETTA BELCASTRO, HELENA FRNDOVA, STEPHEN J. LYE, AND A. KEITH TANSWELL Medical Research Council Group in Lung Development and Lung Biology Programme [R.P.J., X.L., J.C., R.B., H.F., A.K.T.], Hospital for Sick Children Research Institute, the MRC Group in Developmental and Fetal Health, Samuel Lunenfeld Research Institute, Mt. Sinai Hospital [S.J.L.], and the Departments of Obstetrics and Gynaecology [S.J.L.], Paediatrics [A.K.T.] and Physiology [A.K.T., S.J.L.], University of Toronto, Toronto, Ontario, M5S 1A8 Canada. ABSTRACT We hypothesized that reactive O2 species, or their interme- lung cell cultures. We conclude that reactive O2 species, or their diary products, generated during exposure to elevated O2 lead to bioactive intermediaries, are causative in O2-mediated pulmo- pathologic endothelin-1 expression in the newborn lung. Endo- nary hypertension and endothelin-1 up-regulation. It is likely that thelin-1 expression and 8-isoprostane content (an in vivo marker the bioactive lipid peroxidation product, 8-isoprostane, plays a of lipid peroxidation) were examined and found to be elevated key role in pathologic endothelin-1 expression and pulmonary (p Ͻ 0.05) in the lungs of newborn rats with abnormal lung hypertension during oxidant stress. (Pediatr Res 48: 289–298, morphology and pulmonary hypertension, as assessed by right 2000) ventricular hypertrophy, after a 14-d exposure to 60% O2. -

Basic Concepts in Medicinal Chemistry, 2Nd Edition
APP ANSWERS TO CHAPTER QUESTIONS CHAPTER 2 STRUCTURE ANALYSIS CHECKPOINT Checkpoint Drug 1: Venetoclax 1. Answers provided in table below. Functional Group Name Contribution to Water and/or Lipid Solubility A Halogen (chlorine atom) Lipid Solubility B Alicyclic ring, alkyl ring, cycloalkane Lipid Solubility C Tertiary amine (piperazine) Water Solubility D Heterocyclic ring system (pyrrolopyridine) Hydrocarbons: Lipid Solubility Nitrogen atoms: Water Solubility E Aromatic ring; phenyl ring; aromatic hydrocarbon Lipid Solubility F Sulfonamide Water Solubility G Secondary aromatic amine/aniline Water Solubility H Ether Hydrocarbons: Lipid Solubility Oxygen atom: Water Solubility 2. The sulfonamide and tertiary amine will be primarily ionized in most physiological environments and can participate in ion-dipole interactions (as the ion) with water. In the event that they are unionized, they could participate in hydrogen bonding interactions with water. The nitrogen atoms of the heterocyclic ring system, as well as the secondary aromatic amine, and the oxygen atom of the ether will not be appreciably ionized, but can participate in hydrogen bonding interactions with water. Thus, all of these functional groups contribute to the water solubility of venetoclax. The halogen as well as the hydrocarbon chains and rings are not able to ionize or form hydrogen bonds with water and thus contribute to the lipid solubility of venetoclax. 477 Unauthenticated | Downloaded 09/26/21 09:50 PM UTC 478 BASIC CONCEPTS IN MEDICINAL CHEMISTRY 3. Answers provided in table below. Electron Donating or Withdrawing Resonance or Induction A Electron Withdrawing Induction B Both Donates electrons into the aromatic ring through resonance. Withdraws electrons from adjacent methylene groups through induction. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Guadinomines, Type III Secretion System Inhibitors, Produced by Streptomyces Sp
J. Antibiot. 61(4): 222–229, 2008 THE JOURNAL OF ORIGINAL ARTICLE ANTIBIOTICS Guadinomines, Type III Secretion System Inhibitors, Produced by Streptomyces sp. K01-0509 I. Taxonomy, Fermentation, Isolation and Biological Properties Masato Iwatsuki, Ryuji Uchida, Hitomi Yoshijima, Hideaki Ui, Kazuro Shiomi, Atsuko Matsumoto, Yoko Takahashi, Akio Abe, Hiroshi Tomoda, Satoshi O¯ mura Dedicated to the late Prof. Shigeo Iwasaki Received: January 22, 2008 / Accepted: April 4, 2008 © Japan Antibiotics Research Association Abstract Enteropathogenic Escherichia coli (EPEC) Chlamydia spp. [4]. These bacteria use TTSS to deliver expressing the Type III secretion system (TTSS) induced effector proteins into the cytosol of the eukaryotic target hemolysis of sheep blood cells. Using this assay, six cell and depend on their respective TTSS to invade the structurally related compounds designated as guadinomines host, resist phagocytosis, grow in deep tissues, and cause were isolated as inhibitors of TTSS-induced hemolysis by disease [5]. Recent studies have revealed that TTSS is not ion exchange column chromatography and HPLC from the essential for the survival of bacteria and is not found in culture broth of Streptomyces sp. K01-0509. Guadinomines non-pathogenic Gram-negative bacteria except for some A and B showed potent inhibition with IC50 values of 0.02 kinds of symbiotic bacteria. and 0.007 mg/ml, respectively, guadinomine D showed Based on the new concept of “anti-infective drugs” ¯ moderate activity (IC50: 8.5 mg/ml), while guadinomines C1 developed by Omura [6], we have focused on TTSS as a and C2 and guadinomic acid had no activity. new target for anti-infective drugs. -

1 Identification of a Small Molecule Inhibitor of the Aminoglycoside 6'-N
bioRxiv preprint doi: https://doi.org/10.1101/198176; this version posted October 4, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Identification of a Small Molecule Inhibitor of the Aminoglycoside 6'-N- 2 Acetyltransferase Type Ib [AAC(6')-Ib] Using Mixture-Based Combinatorial 3 Libraries 4 5 Tung Tran,1* Kevin Chiem,1* Saumya Jani,1 Brock A. Arivett, 2,3 David Lin,1 Rupali 6 Lad,1 Verónica Jimenez,1 Mary B. Farone, 2 Ginamarie Debevec,4 Radleigh 7 Santos,4 Marc Giulianotti,4 Clemencia Pinilla,5 and Marcelo E. Tolmasky1# 8 9 1Center for Applied Biotechnology Studies, Department of Biological Science, 10 College of Natural Sciences and Mathematics, California State University 11 Fullerton, Fullerton, CA, Departments of 2Biology and 3Chemistry, Middle 12 Tennessee State University, Murfreesboro, TN, 4Torrey Pines Institute for 13 Molecular Studies, Port St. Lucie, FL, and 5Torrey Pines Institute for Molecular 14 Studies, San Diego, CA. 15 16 Word count: 3,879 17 Running Head: Inhibition of AAC(6’)-Ib 18 19 Corresponding Author: Marcelo E. Tolmasky, Professor 20 Mailing address: Department of Biological Science, CNSM, CSUF, PO Box 6850, 21 Fullerton, CA 92831-6850, US. Email: [email protected] 22 23 Co-corresponding Author: Clemencia Pinilla 24 Mailing address: 3550 General Atomics Court, 2-129, San Diego CA 92121-1122, US. 25 Email: [email protected], for or questions related to combinatorial libraries 26 27 *Tung Tran and Kevin Chiem contributed equally to this article 1 bioRxiv preprint doi: https://doi.org/10.1101/198176; this version posted October 4, 2017. -

(12) Patent Application Publication (10) Pub. No.: US 2016/0263257 A1 Elmaleh Et Al
US 20160263257A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0263257 A1 Elmaleh et al. (43) Pub. Date: Sep. 15, 2016 (54) CROMOLYN DERVATIVES AND RELATED (52) U.S. Cl. METHODS OF IMAGING AND TREATMENT CPC ......... A61K 51/0421 (2013.01); A61K 9/4808 (2013.01); A61K 9/0075 (2013.01); A61 K (71) Applicants: David R. ELMALEH, Newton, MA 9/0053 (2013.01); A61K 31/192 (2013.01); (US); Timothy SHOUP, Waltham, MA A61K 31/352 (2013.01); A61K 9/0019 (US) (2013.01) (57) ABSTRACT (72) Inventors: David R. Elmaleh, Newton, MA (US); Novel cromolyn analogs useful as imaging agents for detect Timothy M. Shoup, Waltham, MA ing atherosclerotic plaques and for treating atherosclerosis (US) and Alzheimer's Disease, and methods of making the cro molyn analogs, are disclosed. The cromolyn analogs have (21) Appl. No.: 15/031,098 the general formula: (22) PCT Fed: Oct. 22, 2014 (I) (86) PCT No.: PCT/US1.4f61694 O S 371 (c)(1), (2) Date: Apr. 21, 2016 Related U.S. Application Data (II) (63) Continuation of application No. 14/059.924, filed on Oct. 22, 2013. Publication Classification COH: (51) Int. C. A6 IK 5L/04 (2006.01) wherein X is OH, C-C alkoxyl: Y and Z are independently A6 IK3I/352 (2006.01) selected from a C-C alkyl, C-C alkoxyl, halogen, mi A6 IK3I/92 (2006.01) substituted or C-C substituted amine, F, F, or H; and n A6 IK 9/48 (2006.01) is 1, 2, or 3; and wherein for structure (I), if n are both 1 and A6 IK 9/00 (2006.01) Y and Z are both H and X is OH. -

FDA Listing of Established Pharmacologic Class Text Phrases January 2021
FDA Listing of Established Pharmacologic Class Text Phrases January 2021 FDA EPC Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for Active Moiety Name [indication(s)].” For each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. -

Review of Existing Classification Efforts
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.1.1 Review of existing classification efforts Due date of deliverable: (15.01.2008) Actual submission date: (07.02.2008) Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UGent Revision 1.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public X PP Restricted to other programme participants (including the Commission Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) Task 4.1 : Review of existing classification efforts Authors: Kristof Pil, Elke Raes, Thomas Van den Neste, An-Sofie Goessaert, Jolien Veramme, Alain Verstraete (Ghent University, Belgium) Partners: - F. Javier Alvarez (work package leader), M. Trinidad Gómez-Talegón, Inmaculada Fierro (University of Valladolid, Spain) - Monica Colas, Juan Carlos Gonzalez-Luque (DGT, Spain) - Han de Gier, Sylvia Hummel, Sholeh Mobaser (University of Groningen, the Netherlands) - Martina Albrecht, Michael Heiβing (Bundesanstalt für Straßenwesen, Germany) - Michel Mallaret, Charles Mercier-Guyon (University of Grenoble, Centre Regional de Pharmacovigilance, France) - Vassilis Papakostopoulos, Villy Portouli, Andriani Mousadakou (Centre for Research and Technology Hellas, Greece) DRUID 6th Framework Programme Deliverable D.4.1.1. Revision 1.0 Review of Existing Classification Efforts Page 2 of 127 Introduction DRUID work package 4 focusses on the classification and labeling of medicinal drugs according to their influence on driving performance. -

Interactions of Biocidal Polyhexamethylene Guanidine Hydrochloride and Its Analogs with POPC Model Membranes
polymers Article Interactions of Biocidal Polyhexamethylene Guanidine Hydrochloride and Its Analogs with POPC Model Membranes Xuliang Luo 1,2, Ziran Jiang 1,2, Niya Zhang 1, Zixin Yang 3 and Zhongxin Zhou 1,2,* 1 Key Lab of Agricultural Animal Genetics, Breeding and Reproduction of Ministry of Education, College of Animal Sciences & Technology, Huazhong Agriculture University, 1 Shizishan Street, Wuhan 430070, China; [email protected] (X.L.); [email protected] (Z.J.); [email protected] (N.Z.) 2 The Cooperative Innovation Center for Sustainable Pig Production, Huazhong Agriculture University, 1 Shizishan Street, Wuhan 430070, China 3 College of Sciences, Huazhong Agriculture University, 1 Shizishan Street, Wuhan 430070, China; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +86-27-8728-2091; Fax: +86-27-8728-2091 Received: 14 September 2017; Accepted: 11 October 2017; Published: 17 October 2017 Abstract: The bacterial membrane-targeted polyhexamethylene guanidine hydrochloride (PHGH) and its novel analog polyoctamethylene guanidine hydrochloride (POGH) had excellent antimicrobial activities against antibiotics-resistant bacteria. However, the biocompatibility aspects of PHGH and POGH on the phospholipid membrane of the eukaryotic cell have not yet been considered. Four chemically synthesized cationic oligoguanidine polymers containing alkyl group with different carbon chain lengths, including PHGH, POGH, and their two analogs, were used to determine their interactions