Report of a Germline Double Heterozygote in MSH2 and PALB2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel Breast Cancer ^ Associated BRIP1 (FANCJ/BACH1) Germ- Line Mutation Impairs Protein Stability and Function
Cancer Prevention and Susceptibility A Novel Breast Cancer ^ Associated BRIP1 (FANCJ/BACH1)Germ- line Mutation Impairs Protein Stability and Function Arcangela De Nicolo,1MariellaTancredi,4 Grazia Lombardi,4 Cristina Chantal Flemma,4 Serena Barbuti,4 Claudio Di Cristofano,4 Bijan Sobhian,1Generoso Bevilacqua,4 Ronny Drapkin,2,3 andMariaAdelaideCaligo4 Abstract Purpose: BRCA1-interacting protein 1 (BRIP1; FANCJ/BACH1), which encodes a DNA helicase that interacts with BRCA1, has been suggested to be a low-penetrance breast cancer predispos- ing gene.We aimed to assess whether BRIP1 mutations contribute to breast cancer susceptibility in our population and, if so, to investigate the effect of such mutation(s) on BRIP1function. Experimental Design: A series of49 breast/ovarian cancer families, devoid ofa BRCA1/ BRCA2 mutation, were screened for BRIP1 mutations. Functional analyses, including coimmuno- precipitation and stability assays, were employed to further characterize a previously unreported variant. Results: Five sequence alterations were identified, of which four had been already described. Herein, we report a novel BRIP1 germ-line mutation identified in a woman with early-onset breast cancer. The mutation consists ofa 4-nucleotide deletion (c.2992-2995delAAGA) in BRIP1 exon 20 that causes a shift in the reading frame, disrupts the BRCA1-binding domain of BRIP1, and creates a premature stop codon. Functional analysis ofthe recombinant mutant protein in transfected cells showed that the truncation interferes with the stability of the protein and with its ability to interact with BRCA1. Loss ofthe wild-type BRIP1 allele with retention ofthe mutated one was observed in the patient’s breast tumor tissue. Conclusions: These results, by showing that the newly identified BRIP1 c.2992-2995delAAGA mutation is associated with instability and functional impairment of the encoded protein, provide further evidence of a breast cancer ^ related role for BRIP1. -

Open Full Page
CCR PEDIATRIC ONCOLOGY SERIES CCR Pediatric Oncology Series Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders Michael F. Walsh1, Vivian Y. Chang2, Wendy K. Kohlmann3, Hamish S. Scott4, Christopher Cunniff5, Franck Bourdeaut6, Jan J. Molenaar7, Christopher C. Porter8, John T. Sandlund9, Sharon E. Plon10, Lisa L. Wang10, and Sharon A. Savage11 Abstract DNA repair syndromes are heterogeneous disorders caused by around the world to discuss and develop cancer surveillance pathogenic variants in genes encoding proteins key in DNA guidelines for children with cancer-prone disorders. Herein, replication and/or the cellular response to DNA damage. The we focus on the more common of the rare DNA repair dis- majority of these syndromes are inherited in an autosomal- orders: ataxia telangiectasia, Bloom syndrome, Fanconi ane- recessive manner, but autosomal-dominant and X-linked reces- mia, dyskeratosis congenita, Nijmegen breakage syndrome, sive disorders also exist. The clinical features of patients with DNA Rothmund–Thomson syndrome, and Xeroderma pigmento- repair syndromes are highly varied and dependent on the under- sum. Dedicated syndrome registries and a combination of lying genetic cause. Notably, all patients have elevated risks of basic science and clinical research have led to important in- syndrome-associated cancers, and many of these cancers present sights into the underlying biology of these disorders. Given the in childhood. Although it is clear that the risk of cancer is rarity of these disorders, it is recommended that centralized increased, there are limited data defining the true incidence of centers of excellence be involved directly or through consulta- cancer and almost no evidence-based approaches to cancer tion in caring for patients with heritable DNA repair syn- surveillance in patients with DNA repair disorders. -

Analysis of and Fanconi Anemia Genes in -Negative Spanish Breast Cancer Families María J
Analysis of and Fanconi Anemia genes in -negative Spanish breast cancer families María J. García, Victoria Fernández, Ana Osorio, Alicia Barroso, Gemma Llort, Conxi Lázaro, Ignacio Blanco, Trinidad Caldés, Miguel Hoya, Teresa Ramón y Cajal, et al. To cite this version: María J. García, Victoria Fernández, Ana Osorio, Alicia Barroso, Gemma Llort, et al.. Analysis of and Fanconi Anemia genes in -negative Spanish breast cancer families. Breast Cancer Research and Treatment, Springer Verlag, 2008, 113 (3), pp.545-551. 10.1007/s10549-008-9945-0. hal-00478320 HAL Id: hal-00478320 https://hal.archives-ouvertes.fr/hal-00478320 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Breast Cancer Res Treat (2009) 113:545–551 DOI 10.1007/s10549-008-9945-0 EPIDEMIOLOGY Analysis of FANCB and FANCN/PALB2 Fanconi Anemia genes in BRCA1/2-negative Spanish breast cancer families Marı´a J. Garcı´a Æ Victoria Ferna´ndez Æ Ana Osorio Æ Alicia Barroso Æ Gemma LLort Æ Conxi La´zaro Æ Ignacio Blanco Æ Trinidad Calde´s Æ Miguel de la Hoya Æ Teresa Ramo´n y Cajal Æ Carmen Alonso Æ Marı´a-Isabel Tejada Æ Carlos San Roma´n Æ Luis Robles-Dı´az Æ Miguel Urioste Æ Javier Benı´tez Received: 12 February 2008 / Accepted: 12 February 2008 / Published online: 27 February 2008 Ó Springer Science+Business Media, LLC. -

The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice
G C A T T A C G G C A T genes Review The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice Luigia Stefania Stucci 1,* , Valeria Internò 1 , Marco Tucci 1,2 , Martina Perrone 1, Francesco Mannavola 1 , Raffaele Palmirotta 3 and Camillo Porta 1 1 Division of Medical Oncology, Department of Biomedical Sciences and Human Oncology, University of Bari ‘Aldo Moro’, A.O.U. Consorziale Policlinico di Bari, 70121 Bari, Italy; [email protected] (V.I.); [email protected] (M.T.); [email protected] (M.P.); [email protected] (F.M.); [email protected] (C.P.) 2 National Cancer Research Center, Tumori Institute IRCCS Giovanni Paolo II, 70121 Bari, Italy 3 Interdisciplinary Department of Medicine, Section of Sciences and Technologies of Laboratory Medicine, University of Bari, 70121 Bari, Italy; [email protected] * Correspondence: [email protected] Abstract: Molecular alterations of the Ataxia-telangiectasia (AT) gene are frequently detected in breast cancer (BC), with an incidence ranging up to 40%. The mutated form, the Ataxia-telangiectasia mutated (ATM) gene, is involved in cell cycle control, apoptosis, oxidative stress, and telomere maintenance, and its role as a risk factor for cancer development is well established. Recent studies have confirmed that some variants of ATM are associated with an increased risk of BC development and a worse prognosis. Thus, many patients harboring ATM mutations develop intermediate- and high-grade disease, and there is a higher rate of lymph node metastatic involvement. The evidence concerning a correlation of ATM gene mutations and the efficacy of therapeutic strategies in BC management are controversial. -

About PALB2 Gene Mutations
About PALB2 Gene Mutations About Genes Recommendations Genes are in every cell in our bodies. Genes are made WOMEN of DNA, which gives instructions to cells about how to Starting at age 30: Mammogram and breast MRI every grow and work together. We have two copies of each year (scheduled 6 months apart) gene in each cell—one from our mother and one from our father. When genes work properly, they help stop Some medicines can lower the risk of getting breast cancer from developing. cancer. Surgery to remove both breasts may be an option for some women who have a strong family history of When it works right, the PALB2 gene works together breast cancer. with the BRCA1 and BRCA2 genes to help prevent WOMEN AND MEN cancer. Sometimes changes to the PALB2 gene happen. These changes are called mutations. Mutations can make Screening for pancreatic cancer has benefits and the PALB2 gene stop working and raise the risk for limitations. We do not recommend this screening for certain types of cancer. most people with PALB2 mutations. People who have a PALB2 mutation and a family history of pancreatic Having a mutation in the PALB2 gene makes your risk cancer should ask their doctor or genetic counselor for of getting breast and pancreatic cancers higher than more information. average. The risks for other cancers may also go up with KIDS AND SIBLINGS PALB2 mutations. Researchers are studying the PALB2 gene to understand more. Children and siblings of people with a PALB2 mutation have a 1 in 2 chance of also having the mutation. -

Inherited Variants in BLM and the Risk and Clinical Characteristics of Breast Cancer
cancers Article Inherited Variants in BLM and the Risk and Clinical Characteristics of Breast Cancer Wojciech Klu´zniak 1, Dominika Wokołorczyk 1, Bogna Rusak 1, Tomasz Huzarski 1,2, Aniruddh Kashyap 1, Klaudia Stempa 1 , Helena Rudnicka 1, Anna Jakubowska 1,3 , Marek Szwiec 4, Sylwia Morawska 1, Katarzyna Gliniewicz 1, Karina Mordak 1, Małgorzata Stawicka 2, Joanna Jarkiewicz-Tretyn 5, Magdalena Cechowska 5, Paweł Domagała 6, Tadeusz D˛ebniak 1, Marcin Lener 1, Jacek Gronwald 1, Jan Lubi ´nski 1, Steven A. Narod 7,8, Mohammad R. Akbari 7,8, Cezary Cybulski 1,* and the Polish Hereditary Breast Cancer Consortium y 1 International Hereditary Cancer Center, Department of Genetics and Pathology, Pomeranian Medical University in Szczecin, 71-252 Szczecin, Poland; [email protected] (W.K.); [email protected] (D.W.); [email protected] (B.R.); [email protected] (T.H.); [email protected] (A.K.); [email protected] (K.S.); [email protected] (H.R.); [email protected] (A.J.); [email protected] (S.M.); [email protected] (K.G.); [email protected] (K.M.); [email protected] (T.D.); [email protected] (M.L.); [email protected] (J.G.); [email protected] (J.L.) 2 Department of Clinical Genetics and Pathology, University of Zielona Góra, 65-046 Zielona Góra, Poland; [email protected] 3 Independent Laboratory of Molecular Biology and Genetic Diagnostics, Pomeranian Medical University in Szczecin, 71-252 Szczecin, Poland 4 Department of Surgery and Oncology, University of Zielona Góra, 65-046 Zielona Góra, Poland; [email protected] -

FANCJ Regulates the Stability of FANCD2/FANCI Proteins and Protects Them from Proteasome and Caspase-3 Dependent Degradation
FANCJ regulates the stability of FANCD2/FANCI proteins and protects them from proteasome and caspase-3 dependent degradation Komaraiah Palle, Ph.D. (Kumar) Assistant Professor of Oncologic Sciences Abraham Mitchell Cancer Research Scholar Mitchell Cancer Institute University of South Alabama Outline • Fanconi anemia (FA) pathway • Role of FA pathway in Genome maintenance • FANCJ and FANCD2 functional relationship • FANCJ-mediated DDR in response to Fork-stalling Fanconi Anemia • Rare, inherited blood disorder. • 1:130,000 births Guido Fanconi 1892-1979 • Affects men and women equally. • Affects all racial and ethnic groups – higher incidence in Ashkenazi Jews and Afrikaners Birth Defects Fanconi anemia pathway • FA is a rare chromosome instability syndrome • Autosomal recessive disorder (or X-linked) • Developmental abnormalities • 17 complementation groups identified to date • FA pathway is involved in DNA repair • Increased cancer susceptibility - many patients develop AML - in adults solid tumors Fanconi Anemia is an aplastic anemia FA patients are prone to multiple types of solid tumors • Increased incidence and earlier onset cancers: oral cavity, GI and genital and reproductive tract head and neck breast esophagus skin liver brain Why? FA is a DNA repair disorder • FA caused by mutations in 17 genes: FANCA FANCF FANCM FANCB FANCG/XRCC9 FANCN/PALB2 FANCC FANCI RAD51C/FANCO FANCD1/BRCA2 FANCJ SLX4/FANCP FANCD2 FANCL ERCC2/XPF/FANCQ FANCE BRCA1/FANCS • FA genes function in DNA repair processes • FA patient cells are highly sensitive -

Scaffolding Protein SPIDR/KIAA0146 Connects the Bloom Syndrome Helicase with Homologous Recombination Repair
Scaffolding protein SPIDR/KIAA0146 connects the Bloom syndrome helicase with homologous recombination repair Li Wan1, Jinhua Han1, Ting Liu1, Shunli Dong, Feng Xie, Hongxia Chen, and Jun Huang2 Life Sciences Institute, Zhejiang University, Hangzhou, Zhejiang 310058, China Edited by James E. Cleaver, University of California, San Francisco, CA, and approved February 26, 2013 (received for review December 1, 2012) The Bloom syndrome gene product, BLM, is a member of the highly of the SDSA pathway (6, 7). The ability of BLM to yield non- conserved RecQ family. An emerging concept is the BLM helicase crossover products is thought to play a critical role in the avoidance collaborates with the homologous recombination (HR) machinery to of chromosomal rearrangements during the homolog-directed re- help avoid undesirable HR events and to achieve a high degree of pair of chromosomal lesions. As a result, cells defective for BLM fidelity during the HR reaction. However, exactly how such coordina- exhibit elevated rates of sister chromatid exchange (SCE) (19–21). tion occurs in vivo is poorly understood. Here, we identified a protein Upon the occurrence of DNA damage, BLM is able to form termed SPIDR (scaffolding protein involved in DNA repair) as the link discrete foci, where it colocalizes with other DNA repair proteins between BLM and the HR machinery. SPIDR independently interacts (22, 23). However, mechanistically how BLM is recruited to sites with BLM and RAD51 and promotes the formation of a BLM/RAD51- of DNA damage and how it collaborates with other proteins to containing complex of biological importance. Consistent with its role mediate recombination repair remain largely unexplored. -

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

Challenges in Reporting Pathogenic/Potentially
www.nature.com/scientificreports OPEN Challenges in reporting pathogenic/ potentially pathogenic variants in 94 cancer predisposing genes - in pediatric patients screened with NGS panels Adela Chirita-Emandi 1,2,6*, Nicoleta Andreescu1,2,6, Cristian G. Zimbru1,3, Paul Tutac1,2, Smaranda Arghirescu4,5, Margit Serban5 & Maria Puiu1,2 The beneft of reporting unsolicited fndings in Next Generation Sequencing (NGS) related to cancer genes in children may have implications for family members, nevertheless, could also cause distress. We aimed to retrospectively investigate germline variants in 94 genes implicated in oncogenesis, in patients referred to NGS testing for various rare genetic diseases and reevaluate the utility of reporting diferent classes of pathogenicity. We used in silico prediction software to classify variants and conducted manual review to examine unsolicited fndings frequencies in 145 children with rare diseases, that underwent sequencing - using a 4813 gene panel. The anonymized reanalysis revealed 18250 variants, of which 126 were considered after fltering. Six pathogenic variants (in BRCA1,BMPR1A,FANCA,FANCC,NBN genes) with cancer related phenotype and three unsolicited variants (in BRCA2,PALB2,RAD50 genes) were reported to patients. Additionally, three unsolicited variants in ATR, BLM (in two individuals), and FANCB genes presented potential cancer susceptibility, were not reported to patients. In retrospect, 4.8% (7/145) of individuals in our cohort had unsolicited NGS fndings related to cancer. More eforts are needed to create an updatable consensus in reporting variants in cancer predisposing genes, especially for children. Consent process is crucial to inform of both value and risk of additional genetic information. Next-Generation Sequencing (NGS) for large panels of genes or exomes are increasingly and successfully used in medical management for rare diseases and cancer. -

PALB2, CHEK2 and ATM Rare Variants and Cancer Risk: Data from COGS
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author J Med Genet Manuscript Author . Author manuscript; Manuscript Author available in PMC 2016 December 30. Published in final edited form as: J Med Genet. 2016 December ; 53(12): 800–811. doi:10.1136/jmedgenet-2016-103839. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS A full list of authors and affiliations appears at the end of the article. Abstract Background—The rarity of mutations in PALB2, CHEK2 and ATM make it difficult to estimate precisely associated cancer risks. Population-based family studies have provided evidence that at least some of these mutations are associated with breast cancer risk as high as those associated with rare BRCA2 mutations. We aimed to estimate the relative risks associated with specific rare variants in PALB2, CHEK2 and ATM via a multicentre case-control study. Methods—We genotyped 10 rare mutations using the custom iCOGS array: PALB2 c.1592delT, c.2816T>G and c.3113G>A, CHEK2 c.349A>G, c.538C>T, c.715G>A, c.1036C>T, c.1312G>T, and c.1343T>G and ATM c.7271T>G. We assessed associations with breast cancer risk (42 671 cases and 42 164 controls), as well as prostate (22 301 cases and 22 320 controls) and ovarian (14 542 cases and 23 491 controls) cancer risk, for each variant. Open Access This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. -
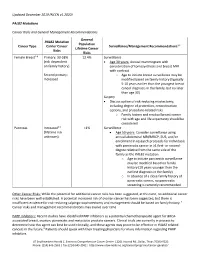
PALB2 Clinical Guide
Updated December 2019 (NCCN v1.2020) PALB2 Mutations Cancer Risks and General Management Recommendations General PALB2 Mutation Population Cancer Type Carrier Cancer Surveillance/Management Recommendations1,2 Lifetime Cancer Risks Risks Female Breast2-8 Primary: 33-58% 12.4% Surveillance (risk dependent Age 30 years: Annual mammogram with on family history) consideration of tomosynthesis and breast MRI with contrast Second primary: o Age to initiate breast surveillance may be Increased modified based on family history (typically 5-10 years earlier than the youngest breast cancer diagnosis in the family, but no later than age 30) Surgery Discuss option of risk-reducing mastectomy, including degree of protection, reconstruction options, and procedure-related risks o Family history and residual breast cancer risk with age and life expectancy should be considered Pancreas Increased3,9 <1% Surveillance (lifetime risk Age 50 years: Consider surveillance using unknown) annual abdominal MRI/MRCP, EUS, and/or enrollment in research protocols for individuals with pancreatic cancer in ≥1 first- or second- degree relative from the same side of the family as the PALB2 mutation o Age to initiate pancreatic surveillance may be modified based on family history (10 years younger than the earliest diagnosis in the family) o In absence of a close family history of pancreatic cancer, no pancreatic screening is currently recommended Other Cancer Risks: While the potential for additional cancer risks has been suggested, at this time, no additional cancer risks have been well established. A potential increased risk of ovarian cancer has been suggested, but there is insufficient evidence for risk-reducing salpingo-oophorectomy and management should be based on family history.