Iodine and Lipase Based Green Oxidation Technology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446
Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, D.C. 20460 OFFICE OF CHEMICAL SAFETY AND POLLUTION PREVENTION MEMORANDUM Date: March 11, 2020 SUBJECT: Registration Review Draft Risk Assessment for the Peroxy Compounds PC Code: 000595, 063201, 063604, 063607, DP Barcode: 455445, 455446 063209, 128860 Decision No: 558073, 558074 Docket No: EPA-HQ-OPP-2009-0546 Regulatory Action: Registration Review Case No: 6059, 4072, 5081 Risk Assessment Type: DRA CAS No: 7722-84-1, 79-21-0, 33734-57-5, 15630-89-4, 10058-23-8, 70693-62-8 TO: Kendall Ziner, Chemical Review Manager Rick Fehir, Ph.D., Team Lead Rose Kyprianou, Branch Chief Regulatory Management Branch (RMB) II Antimicrobials Division (7510P) Office of Pesticide Programs FROM: Andrew Byro, Ph.D., Chemist Kathryn Korthauer, Biologist Timothy Dole, Industrial Hygienist Deborah Burgin, Ph.D., DABT, Toxicologist Risk Assessment and Science Support Branch Antimicrobials Division (7510P) Office of Pesticide Programs THROUGH: Judy Facey, Ph.D., Human Health Risk Assessment Process Leader MP for JF Diana Hsieh, Ecological Risk Assessment Process Leader MP for DH Timothy Leighton, Senior Science Advisor MP for TL Laura Parsons, Associate Branch Chief Melissa Panger, Ph.D., Branch Chief Risk Assessment and Science Support Branch Antimicrobials Division (7510P) This document provides the draft human health and ecological risk assessment conducted in support of the antimicrobial use sites of the following peroxy compounds: hydrogen peroxide, peracetic acid, peroxyoctanoic acid, and sodium percarbonate. Page 1 of 74 Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446 Although the peroxymonosulfate compounds were included in the peroxy compounds Final Work Plan (FWP), they will not be included in this risk assessment. -

A COORDINATION and REDOX CHEMISTRY of ANTIMONY-BASED LIGANDS and APPLICATIONS in ORGANOMETALLIC CATALYSIS a Dissertation by YING
COORDINATION AND REDOX CHEMISTRY OF ANTIMONY-BASED LIGANDS AND APPLICATIONS IN ORGANOMETALLIC CATALYSIS A Dissertation by YING-HAO LO Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, François P. Gabbaï Committee Members, Michael B. Hall Michael Nippe Hung-Jen Wu Head of Department, Simon W. North December 2019 Major Subject: Chemistry Copyright 2019 Ying-Hao Lo a ABSTRACT The use of Z-ligands to modulate the electronic property of transition metal centers is a powerful strategy in catalyst design. Our group has shown that antimony- based ambiphilic ligand, accept electron density from adjacent metal centers by the σ* orbital of the antimony (V) center and thus increasing the electrophilic reactivity of the trancition metal center. In this thesis, we were eager to determine if the charge of the Z- type ligand can be used to further enhance this effect. To this end, we synthesized a 2+ 2+ dicationic gold complex [46] ([(o‐(Ph2P)C6H4)2(o‐Ph2PO)C6H4)SbAuCl] ) featuring a dicationic antimony (V) ligand with a phosphine oxide arm coordinating to the antimony center. Both experimental and computational results show that the gold complex possess a strong Au→Sb interaction reinforced by the dicationic character of the antimony center. The gold‐bound chloride anion of this complex is rather inert and necessitates the addition of excess AgNTf2 to undergo activation. The activated complex, referred to as 2+ 2+ [47] ([((o‐(Ph2P)C6H4)2(o‐Ph2PO)C6H4)SbAuNTf2] ) readily catalyzes both the polymerization and the hydroamination of styrene. -

United States Patent (19) 11) 4,183,924 Green Et Al
United States Patent (19) 11) 4,183,924 Green et al. 45 Jan. 15, 1980 (54) 17a-CHLORO-179-HYDROCARBONSULFI 58) Field of Search .................... 260/239.55 R, 397.3, NYL-1,4-ANDROSTADENES AND THE 260/.397.45; 424/242; Steroids MS File/ CORRESPONDING 176-HYDROCARBONSULFONYL 56 References Cited DERVATIVES AND THEIR USE AS U.S. PATENT DOCUMENTS ANT-ACNE AGENTS 4,091,036 5/1978 Varma ........................... 260/.397.3 X 75 Inventors: Michael J. Green, Kendall Park; Primary Examiner-Ethel G. Love Robert Tiberi, Englishtown, both of Attorney, Agent, or Firm-Elizabeth A. Bellamy; Mary N.J. S. King 73 Assignee: Schering Corporation, Kenilworth, N.J. 57 ABSTRACT Novel 17a-chloro-1743-hydrocarbonsulfinyl and 17a 21) Appl. No.: 881,217 chloro-17 (3-hydrocarbonsulfonyl derivatives of 3-oxo (22 Filed: Feb. 27, 1978 1,4-androstadienes and their use as anti-acne agents are 51) Int. C.? ...................... A61K 31/565; C07J 31/00 described. 52 U.S.C. ........................... 424/242; 260/239.55 R; 260/.397.3; 260/.397.45 18 Claims, No Drawings 4,183,924 1. 2 Preferred compounds of our invention are those de 17a-CHLORO-176-HYDROCARBONSULFINYL fined by the following formula II: 1,4-ANDROSTADIENES AND THE CORRESPONDING II 176-HYDROCARBONSULFONYL DERIVATIVES 5 AND THER USE AS ANT-ACNE AGENTS FIELD OF THE INVENTION This invention relates to novel compositions-of-mat ter, to methods for their manufacture and methods for 10 their use as anti-acne agents. Specifically, this invention relates to novel 17a chloro-176-hydrocarbonsulfinyl and 17a-chloro-1718 hydrocarbonsulfonyl derivatives of 3-oxo-1,4-andros 15 tadienes useful as anti-acne agents. -

Addition Reactions and Photoisomerization of Cis,Trans-1,5-Cyclodecadiene
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1970 Addition Reactions and Photoisomerization of Cis,trans-1,5-Cyclodecadiene. Hsin-hsiong Hsieh Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Hsieh, Hsin-hsiong, "Addition Reactions and Photoisomerization of Cis,trans-1,5-Cyclodecadiene." (1970). LSU Historical Dissertations and Theses. 1859. https://digitalcommons.lsu.edu/gradschool_disstheses/1859 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. 71-6579 HSIEH, Hsin-Hsiong, 1936- ADDITION REACTIONS AND PHOTOISOMERIZATION OF CIS,TRANS-1,5-CYCLODECADIENE. The Louisiana State University and Agricultural and Mechanical College, Ph.D., 1970 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan ADDITION REACTIONS AND PHOTOISOMERIZATION OF CIS,TRANS-1,5“CYCLODECADIENE A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by Hsin-Hsiong Hsieh B.S., Taiwan Provincial Chung-Hsing University, i960 M.S., Louisiana State University, -

WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 3/04 (2006.01) A61K 33/40 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/10 (2006.01) A61K 38/44 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 35/74 (2015.01) A61K 31/17 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 16/034973 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 3 1 May 2016 (3 1.05.2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/169,480 1 June 2015 (01 .06.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/327,283 25 April 2016 (25.04.2016) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: XENO BIOSCIENCES INC. -
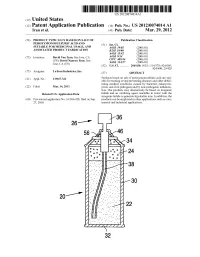
Us 2012/0074.014 A1 2 .. 1
US 2012O074O14A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0074.014 A1 Tran et al. (43) Pub. Date: Mar. 29, 2012 (54) PRODUCT TYPICALLY BASED ON SALT OF Publication Classification PEROXYMONOSULFURIC ACID AND (51) Int. Cl SUITABLE FOR MEDICINAL USAGE, AND iBio/02 (2006.01) ASSOCATED PRODUCT FABRICATION B23P 19/00 (2006.01) A633/42 (2006.01) (75) Inventors: David Van Tran, San Jose, CA A6IR 9/14 (2006.01) (US); David Nguyen Tran, San CD7C 409/244 (2006.01) Jose, CA (US) A6II 3/327 (2006.01) s (52) U.S. Cl. ............. 206/438: 562/1; 514/578; 424/605; 424/400; 29/428 (73) Assignee: LuTran Industries, Inc. (57) ABSTRACT 21) Appl. No.: 13AO47,742 Products based on salt of peroxyperoxVmonosulfuric acid are suit (21) Appl. No 9 able for treating or/and preventing diseases and other debili tating medical conditions caused by bacterial, eukaryotic, (22) Filed: Mar. 14, 2011 prion, and viral pathogens and by non-pathogenic inflamma tion. The products may alternatively be based on inorganic Related U.S. Application Data halide and an oxidizing agent reactable in water with the inorganic halide to generate hypohalite ions. In addition, the (60) Provisional application No. 61/386,928, filed on Sep. products can be employed in other applications such as com 27, 2010. mercial and industrial applications. 5T. 2 .. 2. 1.4. J., Patent Application Publication Mar. 29, 2012 Sheet 1 of 2 US 2012/0074014 A1 cro Q-D 22 Fig. 2a Fig.2b Patent Application Publication Mar. 29, 2012 Sheet 2 of 2 US 2012/0074014 A1 90 92 US 2012/0074014 A1 Mar. -

Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-Free Bleaching of Hardwoods Prehydrolysis-Kraft Pulps
Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps 著者 RIZALUDDIN Andri Taufick year 2016 その他のタイトル 広葉樹材前加水分解クラフトパルプの完全無塩素漂 白のためのモノ過硫酸処理に関する研究 学位授与大学 筑波大学 (University of Tsukuba) 学位授与年度 2015 報告番号 12102甲第7760号 URL http://hdl.handle.net/2241/00143908 Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps January 2016 Andri Taufick RIZALUDDIN Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps A Dissertation Submitted to The Graduate School of Life and Environmental Sciences, The University of Tsukuba In Partial Fulfillment on the Requirements For the Degree of Doctor of Philosophy in Bioresource Engineering (Doctoral Program in Appropriate Technology and Sciences for Sustainable Development) Andri Taufick RIZALUDDIN Table of Contents Chapter 1 Introduction ........................................................................................................... 1 1.1 Indonesian pulp and paper industry........................................................................... 1 1.2 Indonesia’s environmental policies regarding the pulp and paper industry .............. 2 1.3 Bleaching process .................................................................................................... 11 1.3.1 Oxygen bleaching .............................................................................................. 11 1.3.2 Chlorine dioxide bleaching -

Quality Performs
QUALITY PERFORMS. Monopersulfate Compound General Technical Attributes. QUALITY WORKS. MONOPERSULFATE COMPOUND PRODUCT INFORMATION What is OxoneTM? Applications* Oxone™ monopersulfate compound is a white, granular, n Swimming pool shock oxidizer free-flowing peroxygen that provides powerful non-chlorine n Printed wiring board microetchant oxidation for a wide variety of industrial and consumer uses. n Repulping aid for wet-strength resin destruction n Odor control agent in wastewater treatment The active ingredient of Oxone™ is potassium peroxymono- n Bleach component in denture cleanser and laundry sulfate, KHSO5, commonly known as potassium monoper- formulations sulfate, which is present as a component of a triple salt with n Disinfectant active ingredient n the formula 2KHSO5•KHSO4•K2SO4 (pentapotassium bis- Other uses, where its combination of powerful oxidation (peroxymonosulphate) bis(sulphate), [CAS 70693-62-8]). and relative safe handling properties are of value *As with any product, use of Oxone™ in a given application must be tested (including field testing, etc.) by the user in advance to determine suitability. The oxidizing power of Oxone™ is derived from its peracid chemistry; it is the first neutralization salt of peroxymonosulfuric acid H2SO5 (also known as Caro’s acid). Figure 2: Effect of pH on Oxone™ Solution Stability Standard Potential (3 wt% Solution at 32°C [90°F]) 110The standard electrode potential (E°) of KHSO5 is given by 2 the following half cell reaction: 100 20 21C (0F) – + – 0 90HSO5 + 2H + 2e- HSO4 + H2O E = 1.85V 1 0The thermodynamic potential is high enough for many 10 0room temperature oxidations, including: 5 3C (9F) 60 0 n Halide to active halogen pH 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 0n Oxidation of reduced sulfur and nitrogen compounds n Cyanide to cyanate 40 If a decomposition is associated with high temperature, n Epoxidation of olefins decomposition of the constituent salts of Oxone™ may 30n Baeyer-Villiger oxidation of ketones generate sulfuric acid, sulfur dioxide, or sulfur trioxide. -

Tailored Interfaces in Fiber-Reinforced Elastomers: a Surface Treatment Study on Optimized Load Coupling Via the Modified Fiber Bundle Debond Technique
polymers Article Tailored Interfaces in Fiber-Reinforced Elastomers: A Surface Treatment Study on Optimized Load Coupling via the Modified Fiber Bundle Debond Technique Julia Beter 1,* , Boris Maroh 1, Bernd Schrittesser 1 , Inge Mühlbacher 1, Thomas Griesser 2, Sandra Schlögl 1 , Peter Filipp Fuchs 1 and Gerald Pinter 3 1 Polymer Competence Center Leoben GmbH, Roseggerstrasse 12, 8700 Leoben, Austria; [email protected] (B.M.); [email protected] (B.S.); [email protected] (I.M.); [email protected] (S.S.); [email protected] (P.F.F.) 2 Chair of Chemistry of Polymeric Materials, Montanuniversitaet Leoben, Otto-Gloeckel Strasse 2, 8700 Leoben, Austria; [email protected] 3 Department of Polymer Engineering and Science, Montanuniversitaet Leoben, Otto-Gloeckel Strasse 2, 8700 Leoben, Austria; [email protected] * Correspondence: [email protected]; Tel.: +43-3842-42962-31 Abstract: The interface between the reinforcement and surrounding matrix in a fibrous composite is decisive and critical for maintaining component performance, durability, and mechanical structure properties for load coupling assessment, especially for highly flexible composite materials. The clear trend towards tailored solutions reveals that an in-depth knowledge on surface treating methods to enhance the fiber–matrix interfacial interaction and adhesion properties for an optimized load transfer needs to be ensured. This research aims to quantify the effect of several surface treatments for glass fibers applied in endless fiber-reinforced elastomers with pronounced high deformations. Due to this, the glass fiber surface is directly modified with selected sizings, using a wet chemical Citation: Beter, J.; Maroh, B.; treatment, and characterized according to chemical and mechanical aspects. -

Jeffrey E. Lucius U.S. Geological Survey This Report Is Preliminary
U. S. DEPARTMENT OF THE INTERIOR GEOLOGICAL SURVEY PHYSICAL AND CHEMICAL PROPERTIES AND HEALTH EFFECTS OF THIRTY-THREE TOXIC ORGANIC CHEMICALS Jeffrey E. Lucius U.S. Geological Survey Open-File Report 87-428 August 1987 This report is preliminary and has not been reviewed for conformity with U.S. Geological Survey editorial standards. Any use of trade names is for descriptive purposes only and does not imply endorsement by the U.S. Geological Survey. CONTENTS Page Introduction 4 The Properties 6 Abbreviations 13 Conversion Factors 16 Summary Tables 17 Acetic acid 27 Acetone 31 Benzene 34 Bis(2-ethylhexyl)phthalate 37 Bromoform 39 Carbon tetrachloride 42 Chlorobenzene 46 Chloroethane 49 Chloroform 52 Cyclohexane 55 Di-n-butyl phthalate 58 1.1-Dichloroethane 60 1.2-Dichloroethane 63 1,1-Dichloroethene 66 trans-1,2-Dichloroethene 69 Dimethyl sulfoxide 71 1,4-Dioxane 74 Ethanol 77 Ethylbenzene 81 Ethylene dibromide 84 Methanol 87 Methylene chloride 91 Naphthalene 94 Phenol 97 Quinoline 101 Tetrachloroethene 103 Toluene 106 1,1,1-Trichloroethane 109 Trichloroethene 112 Vinyl chloride 115 Water 118 m-Xylene 121 o-Xylene 124 p-Xylene 127 References and Bibliography 130 SUMMARY TABLES page 1. Ranking of top 20 organic ground water contaminants based on number of sites at which each contaminant was detected. 17 2. Selected toxic organic chemicals ordered by Chemical Abstract Service Registry Number (CAS RN). Those chemicals on the U.S. EPA top 100 hazardous substances list are also noted. 18 3. Selected toxic organic chemicals ordered by number of carbon and hydrogen atoms. 19 4. Ranking of selected toxic organic chemicals by specific gravity at room temperature. -

19650003847.Pdf
NATIONAL BUREAU OF STANDARDS REPO RT 8595 PRELIMINARY REPORT ON A SURVEY OF THERMODYNAMIC PROPERTIES OF THE COMPOUNDS OF THE ELEMENTS CHNOPS i N65 2 CACC£S$IO NUMB£:R) CTHRU) ~ ___f£ / I- (PAGES) (CdOEI ::; ~ CIU - d -P?02c2/ :33 (NASA CR OR TMX OR AD NUMBER) (CATEQORY) Progress Report for the Period 1 August to 31 October 1964 to National Aeronautics and Space Administration GPO PRICE $ _____ OTS PRICE(S) $ 1 November 1964 Hard copy (He) of· cJV r Microfiche (MF) __....;... : .:::.j~I?J~_ <@> U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARD S The ational Bureau of Standards is a principal focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. It responsibilities include development and main tena nce of the national stand· ards of measurement, and the provisions of means for making measurements consi tent with those standard ; determination of physical constants and properties of materials; development of methods for testing materials, mechanisms, and structures, and making such tests as may be nece sary, particu larl: for ~overn ment agencies; cooperation in the establi hment of standard practices for incorpora tion in codes and specifications; advisory service to government agencies on scientific and technical problems; invention and development of device to serve special needs of the Government: assi tance to indus! r). business. and consumers in the development and acceptance of commercial tandard and simplified trade practice recommendations; administration of programs in cooperation with United tates bu iness groups and standards organizations for the development of international standard of practice; and maintenance of a clearinghouse for the collection and dis emination of scientific, tech nical. -

Dalton Transactions Accepted Manuscript
Dalton Transactions Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/dalton Page 1 of 12 Dalton Transactions Dalton Transactions RSC Publishing ARTICLE Oxidative Halogenation of Cisplatin and Carboplatin: Synthesis, Spectroscopy, and Crystal and Molecular Structures of Pt(IV) Prodrugs Timothy C. Johnstone a, Sarah M. Alexander a, Justin J. Wilson a, and Stephen J. Cite this: DOI: 10.1039/x0xx00000x Lippard a* A series of Pt(IV) prodrugs has been obtained by oxidative halogenation of either cisplatin or Received 00th January 2012, carboplatin. Iodobenzene dichloride is a general reagent that cleanly provides prodrugs bearing Accepted 00th January 2012 axial chlorides without the need to prepare intervening Pt(IV) intermediates or handle chlorine gas.