Epigenetics and Transcription Regulation During Eukaryotic Diversification: the Saga of TFIID
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METABOLIC EVOLUTION in GALDIERIA SULPHURARIA By
METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA By CHAD M. TERNES Bachelor of Science in Botany Oklahoma State University Stillwater, Oklahoma 2009 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2015 METABOLIC EVOLUTION IN GALDIERIA SUPHURARIA Dissertation Approved: Dr. Gerald Schoenknecht Dissertation Adviser Dr. David Meinke Dr. Andrew Doust Dr. Patricia Canaan ii Name: CHAD M. TERNES Date of Degree: MAY, 2015 Title of Study: METABOLIC EVOLUTION IN GALDIERIA SULPHURARIA Major Field: PLANT SCIENCE Abstract: The thermoacidophilic, unicellular, red alga Galdieria sulphuraria possesses characteristics, including salt and heavy metal tolerance, unsurpassed by any other alga. Like most plastid bearing eukaryotes, G. sulphuraria can grow photoautotrophically. Additionally, it can also grow solely as a heterotroph, which results in the cessation of photosynthetic pigment biosynthesis. The ability to grow heterotrophically is likely correlated with G. sulphuraria ’s broad capacity for carbon metabolism, which rivals that of fungi. Annotation of the metabolic pathways encoded by the genome of G. sulphuraria revealed several pathways that are uncharacteristic for plants and algae, even red algae. Phylogenetic analyses of the enzymes underlying the metabolic pathways suggest multiple instances of horizontal gene transfer, in addition to endosymbiotic gene transfer and conservation through ancestry. Although some metabolic pathways as a whole appear to be retained through ancestry, genes encoding individual enzymes within a pathway were substituted by genes that were acquired horizontally from other domains of life. Thus, metabolic pathways in G. sulphuraria appear to be composed of a ‘metabolic patchwork’, underscored by a mosaic of genes resulting from multiple evolutionary processes. -

Characterization of Two Undescribed Mucoralean Species with Specific
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 26 March 2018 doi:10.20944/preprints201803.0204.v1 1 Article 2 Characterization of Two Undescribed Mucoralean 3 Species with Specific Habitats in Korea 4 Seo Hee Lee, Thuong T. T. Nguyen and Hyang Burm Lee* 5 Division of Food Technology, Biotechnology and Agrochemistry, College of Agriculture and Life Sciences, 6 Chonnam National University, Gwangju 61186, Korea; [email protected] (S.H.L.); 7 [email protected] (T.T.T.N.) 8 * Correspondence: [email protected]; Tel.: +82-(0)62-530-2136 9 10 Abstract: The order Mucorales, the largest in number of species within the Mucoromycotina, 11 comprises typically fast-growing saprotrophic fungi. During a study of the fungal diversity of 12 undiscovered taxa in Korea, two mucoralean strains, CNUFC-GWD3-9 and CNUFC-EGF1-4, were 13 isolated from specific habitats including freshwater and fecal samples, respectively, in Korea. The 14 strains were analyzed both for morphology and phylogeny based on the internal transcribed 15 spacer (ITS) and large subunit (LSU) of 28S ribosomal DNA regions. On the basis of their 16 morphological characteristics and sequence analyses, isolates CNUFC-GWD3-9 and CNUFC- 17 EGF1-4 were confirmed to be Gilbertella persicaria and Pilobolus crystallinus, respectively.To the 18 best of our knowledge, there are no published literature records of these two genera in Korea. 19 Keywords: Gilbertella persicaria; Pilobolus crystallinus; mucoralean fungi; phylogeny; morphology; 20 undiscovered taxa 21 22 1. Introduction 23 Previously, taxa of the former phylum Zygomycota were distributed among the phylum 24 Glomeromycota and four subphyla incertae sedis, including Mucoromycotina, Kickxellomycotina, 25 Zoopagomycotina, and Entomophthoromycotina [1]. -

Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina)
| YEASTBOOK GENOME ORGANIZATION AND INTEGRITY Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina) Bernard A. Dujon*,†,1 and Edward J. Louis‡,§ *Department Genomes and Genetics, Institut Pasteur, Centre National de la Recherche Scientifique UMR3525, 75724-CEDEX15 Paris, France, †University Pierre and Marie Curie UFR927, 75005 Paris, France, ‡Centre for Genetic Architecture of Complex Traits, and xDepartment of Genetics, University of Leicester, LE1 7RH, United Kingdom ORCID ID: 0000-0003-1157-3608 (E.J.L.) ABSTRACT Considerable progress in our understanding of yeast genomes and their evolution has been made over the last decade with the sequencing, analysis, and comparisons of numerous species, strains, or isolates of diverse origins. The role played by yeasts in natural environments as well as in artificial manufactures, combined with the importance of some species as model experimental systems sustained this effort. At the same time, their enormous evolutionary diversity (there are yeast species in every subphylum of Dikarya) sparked curiosity but necessitated further efforts to obtain appropriate reference genomes. Today, yeast genomes have been very informative about basic mechanisms of evolution, speciation, hybridization, domestication, as well as about the molecular machineries underlying them. They are also irreplaceable to investigate in detail the complex relationship between genotypes and phenotypes with both theoretical and practical implications. This review examines these questions at two distinct levels offered by the broad evolutionary range of yeasts: inside the best-studied Saccharomyces species complex, and across the entire and diversified subphylum of Saccharomycotina. While obviously revealing evolutionary histories at different scales, data converge to a remarkably coherent picture in which one can estimate the relative importance of intrinsic genome dynamics, including gene birth and loss, vs. -

Supplementary Table S2: New Taxonomic Assignment of Sequences of Basal Fungal Lineages
Supplementary Table S2: New taxonomic assignment of sequences of basal fungal lineages. Fungal sequences were subjected to BLAST-N analysis and checked for their taxonomic placement in the eukaryotic guide-tree of the SILVA release 111. Sequences were classified depending on combined results from the methods mentioned above as well as literature searches. Accession Name New classification Clustering of the sequence in the Best BLAST-N hit number based on combined results eukaryotic guide tree of SILVA Name Accession number E.value Identity AB191431 Uncultured fungus Chytridiomycota Chytridiomycota Basidiobolus haptosporus AF113413.1 0.0 91 AB191432 Unculltured eukaryote Blastocladiomycota Blastocladiomycota Rhizophlyctis rosea NG_017175.1 0.0 91 AB252775 Uncultured eukaryote Chytridiomycota Chytridiomycota Blastocladiales sp. EF565163.1 0.0 91 AB252776 Uncultured eukaryote Fungi Nucletmycea_Fonticula Rhizophydium sp. AF164270.2 0.0 87 AB252777 Uncultured eukaryote Chytridiomycota Chytridiomycota Basidiobolus haptosporus AF113413.1 0.0 91 AB275063 Uncultured fungus Chytridiomycota Chytridiomycota Catenomyces sp. AY635830.1 0.0 90 AB275064 Uncultured fungus Chytridiomycota Chytridiomycota Endogone lactiflua DQ536471.1 0.0 91 AB433328 Nuclearia thermophila Nuclearia Nucletmycea_Nuclearia Nuclearia thermophila AB433328.1 0.0 100 AB468592 Uncultured fungus Basal clone group I Chytridiomycota Physoderma dulichii DQ536472.1 0.0 90 AB468593 Uncultured fungus Basal clone group I Chytridiomycota Physoderma dulichii DQ536472.1 0.0 91 AB468594 Uncultured -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -
Downloaded (Additional File 1, Table S4)
Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi Capella-Gutiérrez et al. Capella-Gutiérrez et al. BMC Biology 2012, 10:47 http://www.biomedcentral.com/1741-7007/10/47 (31 May 2012) Capella-Gutiérrez et al. BMC Biology 2012, 10:47 http://www.biomedcentral.com/1741-7007/10/47 RESEARCHARTICLE Open Access Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi Salvador Capella-Gutiérrez, Marina Marcet-Houben and Toni Gabaldón* Abstract Background: Microsporidia is one of the taxa that have experienced the most dramatic taxonomic reclassifications. Once thought to be among the earliest diverging eukaryotes, the fungal nature of this group of intracellular pathogens is now widely accepted. However, the specific position of microsporidia within the fungal tree of life is still debated. Due to the presence of accelerated evolutionary rates, phylogenetic analyses involving microsporidia are prone to methodological artifacts, such as long-branch attraction, especially when taxon sampling is limited. Results: Here we exploit the recent availability of six complete microsporidian genomes to re-assess the long- standing question of their phylogenetic position. We show that microsporidians have a similar low level of conservation of gene neighborhood with other groups of fungi when controlling for the confounding effects of recent segmental duplications. A combined analysis of thousands of gene trees supports a topology in which microsporidia is a sister group to all other sequenced fungi. Moreover, this topology received increased support when less informative trees were discarded. This position of microsporidia was also strongly supported based on the combined analysis of 53 concatenated genes, and was robust to filters controlling for rate heterogeneity, compositional bias, long branch attraction and heterotachy. -

The Closest Unicellular Relatives of Animals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Current Biology, Vol. 12, 1773–1778, October 15, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0960-9822(02)01187-9 The Closest Unicellular Relatives of Animals B.F. Lang,1,2 C. O’Kelly,1,3 T. Nerad,4 M.W. Gray,1,5 Results and Discussion and G. Burger1,2,6 1The Canadian Institute for Advanced Research The evolution of the Metazoa from single-celled protists Program in Evolutionary Biology is an issue that has intrigued biologists for more than a 2 De´ partement de Biochimie century. Early morphological and more recent ultra- Universite´ de Montre´ al structural and molecular studies have converged in sup- Succursale Centre-Ville porting the now widely accepted view that animals are Montre´ al, Que´ bec H3C 3J7 related to Fungi, choanoflagellates, and ichthyosporean Canada protists. However, controversy persists as to the spe- 3 Bigelow Laboratory for Ocean Sciences cific evolutionary relationships among these major P.O. Box 475 groups. This uncertainty is reflected in the plethora of 180 McKown Point Road published molecular phylogenies that propose virtually West Boothbay Harbor, Maine 04575 all of the possible alternative tree topologies involving 4 American Type Culture Collection Choanoflagellata, Fungi, Ichthyosporea, and Metazoa. 10801 University Boulevard For example, a monophyletic MetazoaϩChoanoflagel- Manassas, Virginia 20110 lata group has been suggested on the basis of small 5 Department of Biochemistry subunit (SSU) rDNA sequences [1, 6, 7]. Other studies and Molecular Biology using the same sequences have allied Choanoflagellata Dalhousie University with the Fungi [8], placed Choanoflagellata prior to the Halifax, Nova Scotia B3H 4H7 divergence of animals and Fungi [9], or even placed Canada them prior to the divergence of green algae and land plants [10]. -

Sex Is a Ubiquitous, Ancient, and Inherent Attribute of Eukaryotic Life
PAPER Sex is a ubiquitous, ancient, and inherent attribute of COLLOQUIUM eukaryotic life Dave Speijera,1, Julius Lukešb,c, and Marek Eliášd,1 aDepartment of Medical Biochemistry, Academic Medical Center, University of Amsterdam, 1105 AZ, Amsterdam, The Netherlands; bInstitute of Parasitology, Biology Centre, Czech Academy of Sciences, and Faculty of Sciences, University of South Bohemia, 370 05 Ceské Budejovice, Czech Republic; cCanadian Institute for Advanced Research, Toronto, ON, Canada M5G 1Z8; and dDepartment of Biology and Ecology, University of Ostrava, 710 00 Ostrava, Czech Republic Edited by John C. Avise, University of California, Irvine, CA, and approved April 8, 2015 (received for review February 14, 2015) Sexual reproduction and clonality in eukaryotes are mostly Sex in Eukaryotic Microorganisms: More Voyeurs Needed seen as exclusive, the latter being rather exceptional. This view Whereas absence of sex is considered as something scandalous for might be biased by focusing almost exclusively on metazoans. a zoologist, scientists studying protists, which represent the ma- We analyze and discuss reproduction in the context of extant jority of extant eukaryotic diversity (2), are much more ready to eukaryotic diversity, paying special attention to protists. We accept that a particular eukaryotic group has not shown any evi- present results of phylogenetically extended searches for ho- dence of sexual processes. Although sex is very well documented mologs of two proteins functioning in cell and nuclear fusion, in many protist groups, and members of some taxa, such as ciliates respectively (HAP2 and GEX1), providing indirect evidence for (Alveolata), diatoms (Stramenopiles), or green algae (Chlor- these processes in several eukaryotic lineages where sex has oplastida), even serve as models to study various aspects of sex- – not been observed yet. -

University of Oklahoma
UNIVERSITY OF OKLAHOMA GRADUATE COLLEGE MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION SUBMITTED TO THE GRADUATE FACULTY in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY By JOSHUA THOMAS COOPER Norman, Oklahoma 2017 MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION APPROVED FOR THE DEPARTMENT OF MICROBIOLOGY AND PLANT BIOLOGY BY ______________________________ Dr. Boris Wawrik, Chair ______________________________ Dr. J. Phil Gibson ______________________________ Dr. Anne K. Dunn ______________________________ Dr. John Paul Masly ______________________________ Dr. K. David Hambright ii © Copyright by JOSHUA THOMAS COOPER 2017 All Rights Reserved. iii Acknowledgments I would like to thank my two advisors Dr. Boris Wawrik and Dr. J. Phil Gibson for helping me become a better scientist and better educator. I would also like to thank my committee members Dr. Anne K. Dunn, Dr. K. David Hambright, and Dr. J.P. Masly for providing valuable inputs that lead me to carefully consider my research questions. I would also like to thank Dr. J.P. Masly for the opportunity to coauthor a book chapter on the speciation of diatoms. It is still such a privilege that you believed in me and my crazy diatom ideas to form a concise chapter in addition to learn your style of writing has been a benefit to my professional development. I’m also thankful for my first undergraduate research mentor, Dr. Miriam Steinitz-Kannan, now retired from Northern Kentucky University, who was the first to show the amazing wonders of pond scum. Who knew that studying diatoms and algae as an undergraduate would lead me all the way to a Ph.D. -

Are Blastocystis Species Clinically Relevant to Humans?
Are Blastocystis species clinically relevant to humans? Robyn Anne Nagel MB, BS, FRACP A thesis submitted for the degree of Doctor of Philosophy at the University of Queensland in 2015 School of Veterinary Science Title page Culture of human faecal specimen Blastocystis organisms, vacuolated and granular forms, Photographed RAN: x40 magnification, polarised light ii Abstract Blastocystis spp. are the most common enteric parasites found in human stool and yet, the life cycle of the organism is unknown and the clinical relevance uncertain. Robust cysts transmit infection, and many animals carry the parasite. Infection in humans has been linked to Irritable bowel syndrome (IBS). Although Blastocystis carriage is much higher in IBS patients, studies have not been able to confirm Blastocystis spp. are the direct cause of symptoms. Moreover, eradication is often unsuccessful. A number of approaches were utilised in order to investigate the clinical relevance of Blastocystis spp. in human IBS patients. Deconvolutional microscopy with time-lapse imaging and fluorescent spectroscopy of xenic cultures of Blastocystis spp. from IBS patients and healthy individuals was performed. Green autofluorescence (GAF), most prominently in the 557/576 emission spectra, was observed in the vacuolated, granular, amoebic and cystic Blastocystis forms. This first report of GAF in Blastocystis showed that a Blastocystis-specific fluorescein-conjugated antibody could be partially distinguished from GAF. Surface pores of 1m in diameter were observed cyclically opening and closing over 24 hours and may have nutritional or motility functions. Vacuolated forms, extruded a viscous material slowly over 12 hours, a process likely involving osmoregulation. Tear- shaped granules were observed exiting from the surface of an amoebic form but their identity and function could not be elucidated. -

Multigene Eukaryote Phylogeny Reveals the Likely Protozoan Ancestors of Opis- Thokonts (Animals, Fungi, Choanozoans) and Amoebozoa
Accepted Manuscript Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opis- thokonts (animals, fungi, choanozoans) and Amoebozoa Thomas Cavalier-Smith, Ema E. Chao, Elizabeth A. Snell, Cédric Berney, Anna Maria Fiore-Donno, Rhodri Lewis PII: S1055-7903(14)00279-6 DOI: http://dx.doi.org/10.1016/j.ympev.2014.08.012 Reference: YMPEV 4996 To appear in: Molecular Phylogenetics and Evolution Received Date: 24 January 2014 Revised Date: 2 August 2014 Accepted Date: 11 August 2014 Please cite this article as: Cavalier-Smith, T., Chao, E.E., Snell, E.A., Berney, C., Fiore-Donno, A.M., Lewis, R., Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts (animals, fungi, choanozoans) and Amoebozoa, Molecular Phylogenetics and Evolution (2014), doi: http://dx.doi.org/10.1016/ j.ympev.2014.08.012 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 1 Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts 2 (animals, fungi, choanozoans) and Amoebozoa 3 4 Thomas Cavalier-Smith1, Ema E. Chao1, Elizabeth A. Snell1, Cédric Berney1,2, Anna Maria 5 Fiore-Donno1,3, and Rhodri Lewis1 6 7 1Department of Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS, UK. -
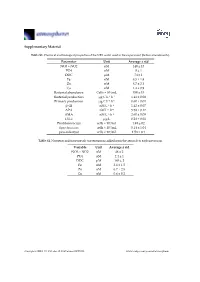
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote