A Rapidly Evolving Polybasic Motif Modulates Bacterial Detection by Guanylate Binding Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Guanine Nucleotidebinding Protein 1 Is One of the Key Molecules
Guanine nucleotide-binding protein 1 is one of the key molecules contributing to cancer cell radioresistance Motoi Fukumoto,1 Tatsuya Amanuma,1 Yoshikazu Kuwahara,1 Tsutomu Shimura,1 Masatoshi Suzuki,1 Shiro Mori,2 Hiroyuki Kumamoto,3 Yohei Saito,4 Yasuhito Ohkubo,4 Zhenfeng Duan,5 Kenji Sano,6 Tomohiro Oguchi,7 Kazuyuki Kainuma,7 Shinichi Usami,7 Kengo Kinoshita,8 Inchul Lee9 and Manabu Fukumoto1 1Department of Pathology, Institute of Development, Aging and Cancer, Tohoku University, Sendai; 2Department of Oral and Maxillofacial Surgery, Tohoku University, Sendai; 3Division of Oral Pathology, Department of Oral Medicine and Surgery, Graduate School of Dentistry, Tohoku University, Sendai; 4Department of Radiopharmacy, Tohoku Pharmaceutical University, Sendai, Japan; 5Department of Hematology/Oncology, Massachusetts General Hospital, Boston, Massachusetts, USA; 6Department of Pathology, School of Medicine, Shinshu University, Matsumoto; 7Department of Otorhinolaryngology, School of Medicine, Shinshu University, Matsumoto, Japan; 8Graduate School of Information Sciences, Tohoku University, Sendai; 9Department of Pathology, Asan Medical Center, Seoul, Korea Key words Standard fractionated radiotherapy for the treatment of cancer consists of daily GTP-binding proteins, head and neck neoplasms, irradiation of 2-Gy X-rays, 5 days a week for 5–8 weeks. To understand the char- neoplasms, radiation, radiation oncology acteristics of radioresistant cancer cells and to develop more effective radiother- Correspondence apy, we established a series of novel, clinically relevant radioresistant (CRR) cells Manabu Fukumoto, Department of Pathology, IDAC, that continue to proliferate with 2-Gy X-ray exposure every 24 h for more than Tohoku University, 4-1 Seiryou-machi, Aoba-ku, Sendai, 30 days in vitro. We studied three human and one murine cell line, and their CRR Japan. -

Contributes to Cell-Autonomous Immunity Against Toxoplasma Gondii Elizabeth M
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2013 Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii Elizabeth M. Selleck Washington University School of Medicine in St. Louis Sarah J. Fentress Washington University School of Medicine in St. Louis Wandy L. Beatty Washington University School of Medicine in St. Louis Daniel Degrandi Heinrich-Heine-Universitat Dusseldorf Klaus Pfeffer Heinrich-Heine-Universitat Dusseldorf See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Selleck, Elizabeth M.; Fentress, Sarah J.; Beatty, Wandy L.; Degrandi, Daniel; Pfeffer, Klaus; Virgin, Herbert W. IV; MacMicking, John D.; and Sibley, L. David, ,"Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii." PLoS Pathogens.,. e1003320. (2013). https://digitalcommons.wustl.edu/open_access_pubs/1496 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Elizabeth M. Selleck, Sarah J. Fentress, Wandy L. Beatty, Daniel Degrandi, Klaus Pfeffer, Herbert W. Virgin IV, John D. MacMicking, and L. David Sibley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1496 Guanylate-binding Protein 1 (Gbp1) Contributes to Cell- autonomous Immunity against Toxoplasma gondii Elizabeth M. Selleck1, Sarah J. Fentress1, Wandy L. Beatty1, Daniel Degrandi2, Klaus Pfeffer2, Herbert W. Virgin IV3, John D. MacMicking4, L. David Sibley1* 1 Department of Molecular Microbiology, Washington University School of Medicine, St. -

Exposing Toxoplasma Gondii Hiding Inside the Vacuole: a Role for Gbps, Autophagy and Host Cell Death
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Curr Opin Manuscript Author Microbiol. Author Manuscript Author manuscript; available in PMC 2020 February 06. Published in final edited form as: Curr Opin Microbiol. 2017 December ; 40: 72–80. doi:10.1016/j.mib.2017.10.021. Exposing Toxoplasma gondii hiding inside the vacuole: a role for GBPs, autophagy and host cell death Jeroen P Saeij1, Eva-Maria Frickel2 1School of Veterinary Medicine, Department of Pathology, Microbiology and Immunology, University of California, Davis, Davis, CA 95616, USA 2The Francis Crick Institute, Host-Toxoplasma Interaction Laboratory, 1 Midland Road, London NW1 1AT, UK Abstract The intracellular parasite Toxoplasma gondii resides inside a vacuole, which shields it from the host’s intracellular defense mechanisms. The cytokine interferon gamma (IFNγ) upregulates host cell effector pathways that are able to destroy the vacuole, restrict parasite growth and induce host cell death. Interferon-inducible GTPases such as the Guanylate Binding Proteins (GBPs), autophagy proteins and ubiquitin-driven mechanisms play important roles in Toxoplasma control in mice and partly also in humans. The host inflammasome is regulated by GBPs in response to bacterial infection in murine cells and may also respond to Toxoplasma infection. Elucidation of murine Toxoplasma defense mechanisms are guiding studies on human cells, while inevitably leading to the discovery of human-specific pathways that often function in a cell type-dependent manner. Introduction Toxoplasma gondii is an important pathogen of animals and humans with ~30% of the world’s population chronically infected. While immunocompetent people generally control the infection, Toxoplasma infection can lead to congenital abnormalities, ocular disease and health problems in the immunocompromised. -

Discovery of a Small Molecule TUBB3/Βiii-Tubulin Modulator in Lung Cancer
Discovery of a small molecule TUBB3/βIII-tubulin modulator in lung cancer Felicity Chao Lin Kao A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy School of Women’s and Children’s Health Faculty of Medicine The University of New South Wales March 2016 TH E UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Kao First name: Felicity Other name/s: Chao Lin Abbreviation for degree as g iven in the University calendar: PhD School: School of Women's and Children's Health Faculty: Faculty of Medicine Title: Discovery of a small molecule TU883/I311 1·tubulin modulator in lung cancer Abstract Non-small Cell Lung Cancer (NSCLC) survival rates are dismal and chemotherapy resistance is a significant clinical problem. 13111-tubulin (encoded by TUBB3 gene) is aberranlly expressed and is associated with chemoresistance and tumour aggressiveness in NSCLC, where it has been identified as a bona fide target for chemosensitisation. Currently, there is no commercially available TUB83J13111-tubulin inhibitor. Regulation of 13111 -tubulin is poorly understood, making it difficult to target this protein. We sought to identify a chemical modulator of TU883J1311Hubulin expression, as it will be a valuable research tool to probe TU883J!3111-tubulin regulation . A novel small molecule TU883/13111-tubulin enhancer, WECC0017371 , was identified in our high throughput screen, based on its ability to modulate TUBB3 promoter activity. WECC001 7371 demonstrated the ability to significantly enhance TUBB3 mRNA and 13111-tubulin protein expression in a time· and dose-dependent manner. Additionally, WECC0017371 did not alter microtubule morphology but enhanced 13111-tubuli n immunostaining in two independent NSCLC cell lines, H460 and H1299, compared to control. -

Detection and Manipulation of Live Antigen-Expressing Cells Using
1 Title: Detection and manipulation of live antigen-expressing cells using 2 conditionally stable nanobodies 3 4 Authors: Jonathan C.Y. Tang1,2†, Eugene Drokhlyansky1,2†, Behzad Etemad3, 5 Stephanie Rudolph4, Binggege Guo1,2, Sui Wang1,2, Emily G, Ellis4, Jonathan Z. Li3, 6 Constance L. Cepko1,2* 7 8 Affiliations: 9 1Howard Hughes Medical Institute 10 2Departments of Genetics and Ophthalmology 11 Harvard Medical School, Boston, MA 02115, USA 12 3Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA 13 4Department of Neurobiology 14 Harvard Medical School, Boston, MA 02115, USA 15 16 †These authors contributed equally to this work 17 *Corresponding author: [email protected] 18 19 Competing Interests: 20 J.C.Y.T, E.D., S.W. and C.L.C. have submitted a U.S. patent application regarding 21 destabilized nanobodies: International Application No. PCT/US2016/027749. Priority: 22 US Prov. Appl. No. 62/148,595. 23 24 25 26 Abstract: The ability to detect and/or manipulate specific cell populations based upon 27 the presence of intracellular protein epitopes would enable many types of studies and 28 applications. Protein binders such as nanobodies (Nbs) can target untagged proteins 29 (antigens) in the intracellular environment. However, genetically expressed protein 30 binders are stable regardless of antigen expression, complicating their use for 31 applications that require cell-specificity. Here, we created a conditional system in which 32 the stability of an Nb depends upon an antigen of interest. We identified Nb framework 33 mutations that can be used to rapidly create destabilized Nbs. -

Microarray Analysis of Novel Genes Involved in HSV- 2 Infection
Microarray analysis of novel genes involved in HSV- 2 infection Hao Zhang Nanjing University of Chinese Medicine Tao Liu ( [email protected] ) Nanjing University of Chinese Medicine https://orcid.org/0000-0002-7654-2995 Research Article Keywords: HSV-2 infection,Microarray analysis,Histospecic gene expression Posted Date: May 12th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-517057/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract Background: Herpes simplex virus type 2 infects the body and becomes an incurable and recurring disease. The pathogenesis of HSV-2 infection is not completely clear. Methods: We analyze the GSE18527 dataset in the GEO database in this paper to obtain distinctively displayed genes(DDGs)in the total sequential RNA of the biopsies of normal and lesioned skin groups, healed skin and lesioned skin groups of genital herpes patients, respectively.The related data of 3 cases of normal skin group, 4 cases of lesioned group and 6 cases of healed group were analyzed.The histospecic gene analysis , functional enrichment and protein interaction network analysis of the differential genes were also performed, and the critical components were selected. Results: 40 up-regulated genes and 43 down-regulated genes were isolated by differential performance assay. Histospecic gene analysis of DDGs suggested that the most abundant system for gene expression was the skin, immune system and the nervous system.Through the construction of core gene combinations, protein interaction network analysis and selection of histospecic distribution genes, 17 associated genes were selected CXCL10,MX1,ISG15,IFIT1,IFIT3,IFIT2,OASL,ISG20,RSAD2,GBP1,IFI44L,DDX58,USP18,CXCL11,GBP5,GBP4 and CXCL9.The above genes are mainly located in the skin, immune system, nervous system and reproductive system. -
Download Validation Data
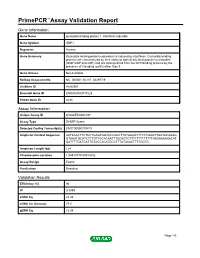
PrimePCR™Assay Validation Report Gene Information Gene Name guanylate binding protein 1, interferon-inducible Gene Symbol GBP1 Organism Human Gene Summary Guanylate binding protein expression is induced by interferon. Guanylate binding proteins are characterized by their ability to specifically bind guanine nucleotides (GMP GDP and GTP) and are distinguished from the GTP-binding proteins by the presence of 2 binding motifs rather than 3. Gene Aliases Not Available RefSeq Accession No. NC_000001.10, NT_032977.9 UniGene ID Hs.62661 Ensembl Gene ID ENSG00000117228 Entrez Gene ID 2633 Assay Information Unique Assay ID qHsaCED0002187 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000370473 Amplicon Context Sequence AATAAACTTCTCCTGAGTGGTACCAGCTTATGAGATTTCCTGAGTTGATACAGAG GTAAATGCATCTTTGTTGCACAATTTACAGTCTTTTTTTTTTTTTAAGAAAAAACAT GATTTTGATCATTGTACCACATGCCTTTATAAACTTTTGGTG Amplicon Length (bp) 124 Chromosome Location 1:89518779-89518932 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 96 R2 0.9999 cDNA Cq 22.48 cDNA Tm (Celsius) 77.5 gDNA Cq 23.04 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report GBP1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

GBP5 Repression Suppresses the Metastatic Potential and PD-L1 Expression in Triple-Negative Breast Cancer
biomedicines Article GBP5 Repression Suppresses the Metastatic Potential and PD-L1 Expression in Triple-Negative Breast Cancer Shun-Wen Cheng 1, Po-Chih Chen 2,3,4 , Min-Hsuan Lin 5, Tzong-Rong Ger 1, Hui-Wen Chiu 5,6,7,* and Yuan-Feng Lin 5,8,* 1 Department of Biomedical Engineering, Chung Yuan Christian University, Taoyuan City 32023, Taiwan; [email protected] (S.-W.C.); [email protected] (T.-R.G.) 2 Neurology Department, Shuang-Ho Hospital, Taipei Medical University, New Taipei City 23561, Taiwan; [email protected] 3 Taipei Neuroscience Institute, Taipei Medical University, New Taipei City 23561, Taiwan 4 Department of Neurology, School of Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan 5 Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan; [email protected] 6 Department of Medical Research, Shuang Ho Hospital, Taipei Medical University, New Taipei City 23561, Taiwan 7 TMU Research Center of Urology and Kidney, Taipei Medical University, Taipei 11031, Taiwan 8 Cell Physiology and Molecular Image Research Center, Wan Fang Hospital, Taipei Medical University, Taipei 11696, Taiwan * Correspondence: [email protected] (H.-W.C.); [email protected] (Y.-F.L.); Tel.: +886-2-22490088 (H.-W.C.); +886-2-2736-1661 (ext. 3106) (Y.-F.L.) Citation: Cheng, S.-W.; Chen, P.-C.; Abstract: Triple-negative breast cancer (TNBC) is the most aggressive breast cancer subtype because Lin, M.-H.; Ger, T.-R.; Chiu, H.-W.; of its high metastatic potential. Immune evasion due to aberrant expression of programmed cell Lin, Y.-F. -

Original Article Nek6 and Hif-1Α Cooperate with the Cytoskeletal Gateway of Drug Resistance to Drive Outcome in Serous Ovarian Cancer
Am J Cancer Res 2015;5(6):1862-1877 www.ajcr.us /ISSN:2156-6976/ajcr0007583 Original Article Nek6 and Hif-1α cooperate with the cytoskeletal gateway of drug resistance to drive outcome in serous ovarian cancer Marta De Donato1, Mara Fanelli2, Marisa Mariani3, Giuseppina Raspaglio1, Deep Pandya3, Shiquan He3, Paul Fiedler3, Marco Petrillo1, Giovanni Scambia1, Cristiano Ferlini3 1Department of Gynecology, Catholic University of The Sacred Heart, Largo Agostino Gemelli 8. 00168 Rome, Italy; 2Laboratory of Molecular Oncology, Jean Paul IInd Research Foundation, Largo Agostino Gemelli 1. 86100 Campobasso, Italy; 3Danbury Hospital Research Institute, 131 West Street 06810 Danbury, CT, USA Received March 4, 2015; Accepted May 10, 2015; Epub May 15, 2015; Published June 1, 2015 Abstract: Hypoxia selects the most aggressive and drug-resistant clones in solid malignancies. One of the pivotal transcription factors induced by hypoxia is Hif-1α. However, in serous ovarian cancer (SEOC), Hif-1α expression is not a prognostic biomarker. This study aims to assess the hypothesis that the serine-threonine kinase Nek6 functions as a downstream effector cooperating with Hif-1α in driving ovarian cancer aggressiveness. Nek6 was overexpressed and Hif-1α was silenced in A2780 cells. Nek6 was also stably silenced in Hey cells. The dependence of Nek6 expression on Hif-1α was assayed as a function of hypoxic growth conditions. Nek6 interaction with the cytoskeletal gateway of drug resistance was investigated with far western blot. The co-expression of NEK6, HIF1A, TUBB3 and GBP1 transcripts was quantified with qPCR in two cohorts of SEOC patients (346 locally treated patients and 344 from the TCGA dataset). -

Structural Mechanism for Guanylate-Binding Proteins (Gbps) Targeting by the Shigella E3 Ligase Ipah9.8
RESEARCH ARTICLE Structural mechanism for guanylate-binding proteins (GBPs) targeting by the Shigella E3 ligase IpaH9.8 1☯ 1☯ 2☯ 1 1 3 Chenggong Ji , Shuo Du , Peng LiID , Qinyu ZhuID , Xiaoke Yang , Chunhong Long , 3 2 1 Jin YuID , Feng ShaoID *, Junyu XiaoID * 1 The State Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China, 2 National Institute of Biological Science (NIBS), Beijing, China, 3 Beijing Computational Science Research Center, Beijing, China a1111111111 a1111111111 ☯ These authors contributed equally to this work. a1111111111 * [email protected] (FS); [email protected] (JX) a1111111111 a1111111111 Abstract The guanylate-binding proteins (GBPs) belong to the dynamin superfamily of GTPases and function in cell-autonomous defense against intracellular pathogens. IpaH9.8, an E3 ligase OPEN ACCESS from the pathogenic bacterium Shigella flexneri, ubiquitinates a subset of GBPs and leads Citation: Ji C, Du S, Li P, Zhu Q, Yang X, Long C, et al. (2019) Structural mechanism for guanylate- to their proteasomal degradation. Here we report the structure of a C-terminally truncated LRR binding proteins (GBPs) targeting by the Shigella GBP1 in complex with the IpaH9.8 Leucine-rich repeat (LRR) domain. IpaH9.8 engages E3 ligase IpaH9.8. PLoS Pathog 15(6): e1007876. the GTPase domain of GBP1, and differences in the Switch II and α3 helix regions render https://doi.org/10.1371/journal.ppat.1007876 some GBPs such as GBP3 and GBP7 resistant to IpaH9.8. Comparisons with other IpaH Editor: Partho Ghosh, University of California San structures uncover interaction hot spots in their LRR domains. -

GBP1 Is a Prognostic Biomarker and Correlates with Immune in Ltrates In
GBP1 Is a Prognostic Biomarker and Correlates With Immune Inltrates in Gastric Cancer Xiaoqiang Feng Center of Stem Cell and Regenerative MedicineGaozhou People’s Hospital Zedan Zhang Shantou University Medical College Qingke Chen Department of Urology, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences) Chujin Ye Department of Urology, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences) Tong Tan Shantou University Medical College Yanlin Tang Shantou University Medical College Hanzhong Chen ( [email protected] ) Department of Urology, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences) Research Article Keywords: GBP1, gastric cancer, prognostic biomarker, tumor immune microenvironment Posted Date: March 1st, 2021 DOI: https://doi.org/10.21203/rs.3.rs-262970/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/25 Abstract Background: The guanylate-binding protein 1 (GBP1) belongs to the member of GTPase dynamin superfamily and plays a major role as tumor suppressor or tumorigen in cancers, such as colon cancer, ovarian cancer, melanoma, and head and neck squamous cell carcinoma. However, the relationship between GBP1 and gastric cancer (GC) remains scanty. Methods: GBP1 mRNA expression in different types of tumors and their corresponding adjacent normal tissues were evaluated via exploring Tumor Immune Estimation Resource (TIMER) and Gene Expression Proling Interactive Analysis (GEPIA) database, while the protein expression was detected by The Human Protein Atlas (HPA). The relationship between the clinical characteristics and outcome of gastric cancer patients was examined through Kaplan–Meier plotter tool. The correlations between GBP1 and tumor immune inltrates were evaluated via TIMER and TISIDB. -

IFN-Inducible Gtpases in Host Cell Defense
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Cell Host & Microbe Review IFN-Inducible GTPases in Host Cell Defense Bae-Hoon Kim,1 Avinash R. Shenoy,1 Pradeep Kumar,1 Clinton J. Bradfield,1 and John D. MacMicking1,* 1Department of Microbial Pathogenesis, Boyer Centre for Molecular Medicine, Yale University School of Medicine, New Haven, CT 06510, USA *Correspondence: [email protected] http://dx.doi.org/10.1016/j.chom.2012.09.007 From plants to humans, the ability to control infection at the level of an individual cell—a process termed cell- autonomous immunity—equates firmly with survival of the species. Recent work has begun to unravel this programmed cell-intrinsic response and the central roles played by IFN-inducible GTPases in defending the mammalian cell’s interior against a diverse group of invading pathogens. These immune GTPases regu- late vesicular traffic and protein complex assembly to stimulate oxidative, autophagic, membranolytic, and inflammasome-related antimicrobial activities within the cytosol, as well as on pathogen-containing vacuoles. Moreover, human genome-wide association studies and disease-related transcriptional profiling have linked mutations in the Immunity-Related GTPase M (IRGM) locus and altered expression of guanylate binding proteins (GBPs) with tuberculosis susceptibility and Crohn’s colitis. Introduction IFNs serve as powerful signals for marshaling host defense Essentially all diploid cells are endowed with a capacity for self throughout the vertebrate kingdom. These cytokines induce defense. This type of host resistance, called cell-autonomous numerous gene families that encode effectors and associated immunity, is ubiquitous throughout both plant and animal king- regulators to direct each protein’s microbicidal or static activity doms (Beutler et al., 2006).