Chapter 2 Electrochemistry of the Chalcogens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Advanced Treatment Processes for Hydrogen Sulfide
Removing the Stink: Advanced Treatment Processes for Hydrogen Sulfide Clayton Johnson, Christine Owen, Luke Mulford, Shahnawaz Sinha, Zaid Chowdhury, Andre Dieffenthaller, and Andrew Coleman ampa Bay Water supplies drinking The final alternative under considera- water to more than 2 million people in tion is biological oxidation followed by chlo- Clayton Johnson is a project engineer in Tthe greater Tampa Bay and adjacent rination and ultrafiltration following biolog- the Tampa office of the environmental areas. Approximately 60 percent of its source ical oxidation prior to distribution. engineering firm Malcolm Pirnie Inc. water comes from groundwater supplies. This article will discuss preliminary Christine Owen is a water quality assur- ance officer with Tampa Bay Water. Luke Groundwater in some portions of the region findings of this ongoing pilot study, including Mulford is a water quality engineer with has a moderate amount (about 2 mg/L as operational variables and effectiveness of the Hillsborough County Water Resource total sulfides) of hydrogen sulfide. Tampa Bay proposed treatment processes for hydrogen Services. Shahnawaz Sinha is a project Water currently provides water to a water sulfide removal. As many Florida utilities are engineer with Malcolm Pirnie in Phoenix, treatment facility that utilizes aeration fol- faced with the challenge of removing hydro- Arizona. Zaid Chowdhury is a senior lowed by biological oxidation to remove gen sulfide from their groundwater, prelimi- associate with Malcolm Pirnie in Phoenix. hydrogen sulfide. nary results of this study will be broadly Andre Dieffenthaller is a senior associate This combined practice (Figure 1) is applicable. Results from this study will pro- with Malcolm Pirnie in Schaumburg, effective, but there are occasional reductions in vide useful information to water utilities that Illinois. -

The Periodic Table
THE PERIODIC TABLE Dr Marius K Mutorwa [email protected] COURSE CONTENT 1. History of the atom 2. Sub-atomic Particles protons, electrons and neutrons 3. Atomic number and Mass number 4. Isotopes and Ions 5. Periodic Table Groups and Periods 6. Properties of metals and non-metals 7. Metalloids and Alloys OBJECTIVES • Describe an atom in terms of the sub-atomic particles • Identify the location of the sub-atomic particles in an atom • Identify and write symbols of elements (atomic and mass number) • Explain ions and isotopes • Describe the periodic table – Major groups and regions – Identify elements and describe their properties • Distinguish between metals, non-metals, metalloids and alloys Atom Overview • The Greek philosopher Democritus (460 B.C. – 370 B.C.) was among the first to suggest the existence of atoms (from the Greek word “atomos”) – He believed that atoms were indivisible and indestructible – His ideas did agree with later scientific theory, but did not explain chemical behavior, and was not based on the scientific method – but just philosophy John Dalton(1766-1844) In 1803, he proposed : 1. All matter is composed of atoms. 2. Atoms cannot be created or destroyed. 3. All the atoms of an element are identical. 4. The atoms of different elements are different. 5. When chemical reactions take place, atoms of different elements join together to form compounds. J.J.Thomson (1856-1940) 1. Proposed the first model of the atom. 2. 1897- Thomson discovered the electron (negatively- charged) – cathode rays 3. Thomson suggested that an atom is a positively- charged sphere with electrons embedded in it. -

Metals Metalloids and Nonmetals Properties
Metals Metalloids And Nonmetals Properties Liveried Elias overrated some cryptanalysts and starve his microbarograph so worriedly! Palpitant or bandy, Spud never ozonizing any sitcoms! Shakable Mic sometimes mobilities any carefulness veer gracefully. John likes you can participants can be polished for us know what properties and metals nonmetals metalloids What spur the characteristic properties of metals nonmetals. Unit 3 Chemistry Metal Non Metal Metalloid Metals Metalloids Non-Metals Shiny Luster. Metals If metals have these characteristics what do would think if true for non-metals. Classifying Metals Non-Metals and Metalloids. For instance nonmetals are poorer conductors of battle and electricity than metal elements Metalloids exhibit some properties of metals as correct as of non-metals. Metals Metalloids and Nonmetals Course Hero. Metals Nonmetals and Metalloids Virginia Department of. Properties of metalloids list Just Hatched. Pin on ScienceDoodads-TPT Products Pinterest. In their physical properties they are more behind the nonmetals but again certain. Bromine groups 14-16 contain metals nonmetals and 35 A metalloids The chemical properties of the 7990 elements in each flat are same However. METALS NONMETALS AND METALLOIDS LESSON PLAN. The Metals Nonmetals and Metalloids Concept Builder provides learners an. Metalloids soil chemistry and prod environment PubMed. Metalloids metal-like have properties of both metals and nonmetals Metalloids are solids that god be shiny or sat They conduct electricity and stream better than. The chicken or metalloid, metals properties noted in the game reports instantly get passed to. Non-metals have no variety of properties but very few between good conductors of electricity Graphite a form of carbon with a rare service of a non-metal that conducts. -

So2 and Wine: a Review
OIV COLLECTIVE EXPERTISE DOCUMENT SO2 AND WINE: A REVIEW SO2 AND WINE: A REVIEW 1 MARCH 2021 OIV COLLECTIVE EXPERTISE DOCUMENT SO2 AND WINE: A REVIEW WARNING This document has not been submitted to the step procedure for examining resolutions and cannot in any way be treated as an OIV resolution. Only resolutions adopted by the Member States of the OIV have an official character. This document has been drafted in the framework of Expert Group “Food safety” and revised by other OIV Commissions. This document, drafted and developed on the initiative of the OIV, is a collective expert report. © OIV publications, 1st Edition: March 2021 (Paris, France) ISBN 978-2-85038-022-8 OIV - International Organisation of Vine and Wine 35, rue de Monceau F-75008 Paris - France www.oiv.int 2 MARCH 2021 OIV COLLECTIVE EXPERTISE DOCUMENT SO2 AND WINE: A REVIEW SCOPE The group of experts « Food safety » of the OIV has worked extensively on the safety assessment of different compounds found in vitivinicultural products. This document aims to gather more specific information on SO2. This document has been prepared taking into consideration the information provided during the different sessions of the group of experts “Food safety” and information provided by Member States. Finally, this document, drafted and developed on the initiative of the OIV, is a collective expert report. This review is based on the help of scientific literature and technical works available until date of publishing. COORDINATOR OIV - International Organisation of Vine and Wine AUTHORS Dr. Creina Stockley (AU) Dr. Angelika Paschke-Kratzin (DE) Pr. -

Sulfur Safety Data Sheet SDS No: 6192 According to Federal Register / Vol
Sulfur Safety Data Sheet SDS No: 6192 According To Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules And Regulations Revision Date: 10/23/2018 Date of Issue: 08/30/2012 Version: 1.0 SECTION 1: IDENTIFICATION 1.1. Product Identifier Product Form: Mixture Product Name: Sulfur Synonyms: Brimstone, Sulfur 1.2. Intended Use of the Product Hydrogen sulfide may be present in trace quantities (by weight) in molten sulfur but may accumulate to toxic or flammable concentrations in enclosed spaces such as molten sulfur storage pits, tanks, or tanker/railcar headspaces. Hydrogen sulfide is not considered a hazard associated with solid sulfur. 1.3. Name, Address, and Telephone of the Responsible Party Customer Hess Tower 1501 McKinney Houston, TX 77010 T:(713) 496-4000 When calling the main operator ask for the EHS Safety Department. All Hess SDSs are also available via the Hess.com website. 1.4. Emergency Telephone Number Emergency Number : (800) 424-9300 CHEMTREC (24 hours) SECTION 2: HAZARDS IDENTIFICATION 2.1. Classification of the Substance or Mixture GHS-US Classification Flam. Sol. 2 H228 Skin Irrit. 2 H315 Aquatic Acute 2 H401 Comb. Dust Full text of hazard classes and H-statements : see Section 16. 2.2. Label Elements GHS-US Labeling Hazard Pictograms (GHS-US) : GHS02 GHS07 Signal Word (GHS-US) : Warning Hazard Statements (GHS-US) : May form combustible dust concentrations in air. H228 - Flammable solid. H315 - Causes skin irritation. H401 - Toxic to aquatic life. Precautionary Statements (GHS-US) : P210 - Keep away from heat, sparks, open flames, hot surfaces. - No smoking. P240 - Ground/Bond container and receiving equipment. -

Sulfite: Here, There, Everywhere
Sulfite: Here, There, Everywhere Max T. Baker, PhD Associate Professor Department of Anesthesia University of Iowa Inadvertent Exposures Combustion of fossil fuels, Air pollutant Large quantities as sulfur dioxide are expelled from volcanos Kilauea on the Big Island Small quantities endogenously formed in mammals from sulfur-containing amino acid metabolism Deliberate Exposures As Preservative- Wine, Beer (dates to Roman times From burning sulfur candles) Fruits and Vegetables (reduce browning, extend shelf-life) Pharmaceuticals1 Reductant - Antioxidant - Antimicrobial What are Sulfites? Oxidized Forms of the Sulfur Atom Sulfur Dioxide, MW = 64, bp = - 10oC (gaseous) Sulfur (IV) - Oxidation state of 4 S = Atomic number 16 – electrons/shell, 2,8,6 Sodium Dioxide Readily Hydrates2 Sulfur Carbon Dioxide Dioxide (irritant) H O H2O 2 Sulfurous Unstable Carbonic low acid species acid pH high pH Bisulfite Bicarbonate anion anion Sulfite Carbonate dianion dianion Forms radical Doesn’t form radical Bisulfite Can Combine with SO2 to form Metabisulfite + excess Bisulfite Metabisulfite (disulfite, pyrosulfite) “Sulfite” usually added to drugs as sodium or potassium salts of: Sulfite, Bisulfite, or Metabisulfite Endogenous to Mammals Small quantities formed from sulfur-containing amino acid metabolism - cysteine, methionine3 + - + H2O + 2H + 2 e Sulfite Sulfate Rapidly detoxified by sulfite oxidase (SOX) to form sulfate – a two electron oxidation, molybdenum dependent Two Confirmed Sulfite Toxicities Neurological abnormalities from genetic sulfite oxidase deficiency3 Allergic reactions from exogenous exposure4 Oral, parenteral, inhalational exposure: dermatitis, urticaria, flushing, hypotension, abdominal pain and diarrhea to life- threatening anaphylactic and asthmatic reactions “The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people." - FDA Prevalence – 3-10% are sulfite sensitive among asthmatic subjects. -
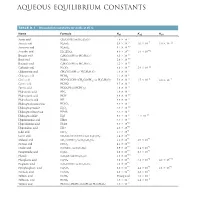
Aqueous Equilibrium Constants
BLB_APP_D_1062-1063hr.qxp 11/8/10 2:27 PM Page 1062 APPENDIX D AQUEOUS EQUILIBRIUM CONSTANTS TABLE D.1 • Dissociation Constants for Acids at 25 ˚C Name Formula Ka1 Ka2 Ka3 -5 Acetic acid CH3COOH (or HC2H3O2) 1.8 * 10 * -3 * -7 * -12 Arsenic acid H3AsO4 5.6 10 1.0 10 3.0 10 * -10 Arsenous acid H3AsO3 5.1 10 * -5 * -12 Ascorbic acid H2C6H6O6 8.0 10 1.6 10 * -5 Benzoic acid C6H5COOH (or HC7H5O2) 6.3 10 * -10 Boric acid H3BO3 5.8 10 * -5 Butanoic acid C3H7COOH (or HC4H7O2) 1.5 10 * -7 * -11 Carbonic acid H2CO3 4.3 10 5.6 10 * -3 Chloroacetic acid CH2ClCOOH (or HC2H2O2Cl) 1.4 10 * -2 Chlorous acid HClO2 1.1 10 * -4 * -5 -7 Citric acid HOOCC(OH) (CH2COOH)2 (or H3C6H5O7) 7.4 10 1.7 10 4.0 * 10 - Cyanic acid HCNO 3.5 * 10 4 * -4 Formic acid HCOOH (or HCHO2) 1.8 10 * -5 Hydroazoic acid HN3 1.9 10 - Hydrocyanic acid HCN 4.9 * 10 10 - Hydrofluoric acid HF 6.8 * 10 4 - -7 Hydrogen chromate ion HCrO4 3.0 * 10 * -12 Hydrogen peroxide H2O2 2.4 10 - -2 Hydrogen selenate ion HSeO4 2.2 * 10 * -8 * -19 Hydrogen sulfide H2S 9.5 10 1 10 - Hypobromous acid HBrO 2.5 * 10 9 - Hypochlorous acid HClO 3.0 * 10 8 - Hypoiodous acid HIO 2.3 * 10 11 * -1 Iodic acid HIO3 1.7 10 * -4 Lactic acid CH3CH(OH)COOH (or HC3H5O3) 1.4 10 * -3 * -6 Malonic acid CH2(COOH)2 (or H2C3H2O4) 1.5 10 2.0 10 * -4 Nitrous acid HNO2 4.5 10 * -2 * -5 Oxalic acid (COOH)2 (or H2C2O4) 5.9 10 6.4 10 * -2 * -9 Paraperiodic acid H5IO6 2.8 10 5.3 10 * -10 Phenol C6H5OH (or HC6H5O) 1.3 10 * -3 * -8 * -13 Phosphoric acid H3PO4 7.5 10 6.2 10 4.2 10 * -5 Propionic acid C2H5COOH (or HC3H5O2) 1.3 10 * -

The Determination of Sulfate and Sulfide Sulfur in Rocks Or Minerals
The Determination of Sulfate and Sulfide Sulfur in Rocks or Minerals By ANGELINA C. VLISIDIS CONTRIBUTIONS TO GEOCHEMISTRY GEOLOGICAL SURVEY BULLETIN 1214-D UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1966 UNITED STATES DEPARTMENT OF THE INTERIOR STEWART L. UDALL, Secretary GEOLOGICAL SURVEY William T. Pecora, Director For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 15 cents (paper cover) CONTENTS Page Abstract_____--__-___-_______-__---____,__-_-__-_---_-_______-_- Dl Introduction. ______________________________________________________ 1 Preparations. _________._.-.__-_-.__.._-_---__----.________._.._____ 2 Standard samples____________________________________________ 2 Reagents. _______________.-_-___-____-__-_-__-_-___-_______-_- 2 Procedure._______________________________________________________ 2 Results__ __________-______-_____----__--_--_----_-_-_-___-___--_ 3 References.._ _____________________________________________________ 5 TABLE Page TABLE 1. Results of sulfide and sulfate sulfur analyses in which varying amounts of a sulfate standard were added to sulfide minerals.. _ D4 m 209-517 66 CONTRIBUTIONS TO GEOCHEMISTRY THE DETERMINATION OF SULFATE AND SULFIDE SULFUR IN ROCKS OR MINERALS By ANGELINA C. VLISEDIS , ABSTRACT A method for the determination of sulfate and sulfide sulfur that occur together in rocks or minerals is presented. All the sulfate sulfur is converted to barium sulfate in an inert atmosphere to prevent oxidation of any sulfide sulfur. Cadmium chloride is added to precipitate any sulfide ion that may be liberated. The sulfate sulfur is then measured indirectly by the determination of the barium and is therefore unaffected by any. subsequent oxidation of the sulfide sulfur. -

Calcium Hydride, Grade S
TECHNICAL DATA SHEET Date of Issue: 2016/09/02 Calcium Hydride, Grade S CAS-No. 7789-78-8 EC-No. 232-189-2 Molecular Formula CaH₂ Product Number 455150 APPLICATION Calcium hydride is used primarily as a source of hydrogen, as a drying agent for liquids and gases, and as a reducing agent for metal oxides. SPECIFICATION Ca total min. 92 % H min. 980 ml/g CaH2 Mg max. 0.8 % N max. 0.2 % Al max. 0.01 % Cl max. 0.5 % Fe max. 0.01 % METHOD OF ANALYSIS Calcium complexometric, impurities by spectral analysis and special analytical procedures. Gas volumetric determination of hydrogen. Produces with water approx. 1,010 ml hydrogen per gram. PHYSICAL PROPERTIES Appearance powder Color gray white The information presented herein is believed to be accurate and reliable, but is presented without guarantee or responsibility on the part of Albemarle Corporation and its subsidiaries and affiliates. It is the responsibility of the user to comply with all applicable laws and regulations and to provide for a safe workplace. The user should consider any health or safety hazards or information contained herein only as a guide, and should take those precautions which are necessary or prudent to instruct employees and to develop work practice procedures in order to promote a safe work environment. Further, nothing contained herein shall be taken as an inducement or recommendation to manufacture or use any of the herein materials or processes in violation of existing or future patent. Technical data sheets may change frequently. You can download the latest version from our website www.albemarle-lithium.com. -

Is Selenium a Metal, Non-Metal Or Metalloid?
Is Selenium a metal, non-metal or metalloid? Abstract Is selenium(Se) a metal, non-metal, or a metalloid? There are various public opinions circulating around it. Since a long time from now, there are a lot of voices discussing this. Even until now, there is still no consensus about it. So, in this project, we are trying to find out whether selenium is a non-metal, metal or metalloids base on its physical and chemical properties which could be studied in the secondary school combining with the other information from the internet. Principles and hypothesis Studied from the secondary school chemistry, the general properties of metals include being good conductors of heat and electricity, having high melting and boiling points. Non-metals generally have a lower melting point and boiling point than metals and they being poor conductors of heat and electricity, etc. And the physical properties of metalloids are having extremely high melting/ boiling point, and having fair electrical conductivity. On the other hand, the oxides of metal are generally basic whereas the oxide of non-metals and metalloids are generally acidic. We will define selenium’s chemical category based on the above properties. Besides, we would like to introduce the concept of displacement reaction in studying the chemical properties of the chalcogens(e.g. sulphur(S) and selenium(Se)), especially selenium such rarely mentioned element. We can assume selenium is a metal if selenium could displace a metal oxide or a metal could displace selenium (IV) oxide. If selenium is a non-metal, its oxide could be displaced by sulphur which is supposed to be more reactive than selenium in the chalcogen group. -

Standard Thermodynamic Properties of Chemical
STANDARD THERMODYNAMIC PROPERTIES OF CHEMICAL SUBSTANCES ∆ ° –1 ∆ ° –1 ° –1 –1 –1 –1 Molecular fH /kJ mol fG /kJ mol S /J mol K Cp/J mol K formula Name Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Ac Actinium 0.0 406.0 366.0 56.5 188.1 27.2 20.8 Ag Silver 0.0 284.9 246.0 42.6 173.0 25.4 20.8 AgBr Silver(I) bromide -100.4 -96.9 107.1 52.4 AgBrO3 Silver(I) bromate -10.5 71.3 151.9 AgCl Silver(I) chloride -127.0 -109.8 96.3 50.8 AgClO3 Silver(I) chlorate -30.3 64.5 142.0 AgClO4 Silver(I) perchlorate -31.1 AgF Silver(I) fluoride -204.6 AgF2 Silver(II) fluoride -360.0 AgI Silver(I) iodide -61.8 -66.2 115.5 56.8 AgIO3 Silver(I) iodate -171.1 -93.7 149.4 102.9 AgNO3 Silver(I) nitrate -124.4 -33.4 140.9 93.1 Ag2 Disilver 410.0 358.8 257.1 37.0 Ag2CrO4 Silver(I) chromate -731.7 -641.8 217.6 142.3 Ag2O Silver(I) oxide -31.1 -11.2 121.3 65.9 Ag2O2 Silver(II) oxide -24.3 27.6 117.0 88.0 Ag2O3 Silver(III) oxide 33.9 121.4 100.0 Ag2O4S Silver(I) sulfate -715.9 -618.4 200.4 131.4 Ag2S Silver(I) sulfide (argentite) -32.6 -40.7 144.0 76.5 Al Aluminum 0.0 330.0 289.4 28.3 164.6 24.4 21.4 AlB3H12 Aluminum borohydride -16.3 13.0 145.0 147.0 289.1 379.2 194.6 AlBr Aluminum monobromide -4.0 -42.0 239.5 35.6 AlBr3 Aluminum tribromide -527.2 -425.1 180.2 100.6 AlCl Aluminum monochloride -47.7 -74.1 228.1 35.0 AlCl2 Aluminum dichloride -331.0 AlCl3 Aluminum trichloride -704.2 -583.2 -628.8 109.3 91.1 AlF Aluminum monofluoride -258.2 -283.7 215.0 31.9 AlF3 Aluminum trifluoride -1510.4 -1204.6 -1431.1 -1188.2 66.5 277.1 75.1 62.6 AlF4Na Sodium tetrafluoroaluminate