Atomic and Molecular Orbitals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Atomic Orbitals
Visualization of Atomic Orbitals Source: Robert R. Gotwals, Jr., Department of Chemistry, North Carolina School of Science and Mathematics, Durham, NC, 27705. Email: [email protected]. Copyright ©2007, all rights reserved, permission to use for non-commercial educational purposes is granted. Introductory Readings Where are the electrons in an atom? There are a number of Models - a description of models that look to describe the location and behavior of some physical object or electrons in atoms. It is important to know this information, as behavior in nature. Models it helps chemists to make statements and predictions about are often mathematical, describing the behavior of how atoms will react with other atoms to form molecules. The some event. model one chooses will oftentimes influence the types of statements one can make about the behavior – that is, the reactivity – of an atom or group of atoms. Bohr model - states that no One model, the Bohr model, describes electrons as small two electrons in an atom can billiard balls, rotating around the positively charged nucleus, have the same set of four much in the way that planets quantum numbers. orbit around the Sun. The placement and motion of electrons are found in fixed and quantifiable levels called orbits, Orbits- where electrons are or, in older terminology, shells. believed to be located in the There are specific numbers of Bohr model. Orbits have a electrons that can be found in fixed amount of energy, and are identified with the each orbit – 2 for the first orbit, principle quantum number 8 for the second, and so forth. -

Chemistry 2000 Slide Set 1: Introduction to the Molecular Orbital Theory
Chemistry 2000 Slide Set 1: Introduction to the molecular orbital theory Marc R. Roussel January 2, 2020 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 1 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms The hydrogenic atom (one nucleus, one electron) is exactly solvable. The solutions of this problem are called atomic orbitals. The square of the orbital wavefunction gives a probability density for the electron, i.e. the probability per unit volume of finding the electron near a particular point in space. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 2 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms (continued) Orbital shapes: 1s 2p 3dx2−y 2 3dz2 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 3 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Multielectron atoms Consider He, the simplest multielectron atom: Electron-electron repulsion makes it impossible to solve for the electronic wavefunctions exactly. A fourth quantum number, ms , which is associated with a new type of angular momentum called spin, enters into the theory. 1 1 For electrons, ms = 2 or − 2 . Pauli exclusion principle: No two electrons can have identical sets of quantum numbers. Consequence: Only two electrons can occupy an orbital. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 4 / 24 The hydrogen molecular ion The quantum mechanics of molecules + H2 is the simplest possible molecule: two nuclei one electron Three-body problem: no exact solutions However, the nuclei are more than 1800 time heavier than the electron, so the electron moves much faster than the nuclei. -

Theoretical Methods That Help Understanding the Structure and Reactivity of Gas Phase Ions
International Journal of Mass Spectrometry 240 (2005) 37–99 Review Theoretical methods that help understanding the structure and reactivity of gas phase ions J.M. Merceroa, J.M. Matxaina, X. Lopeza, D.M. Yorkb, A. Largoc, L.A. Erikssond,e, J.M. Ugaldea,∗ a Kimika Fakultatea, Euskal Herriko Unibertsitatea, P.K. 1072, 20080 Donostia, Euskadi, Spain b Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455-0431, USA c Departamento de Qu´ımica-F´ısica, Universidad de Valladolid, Prado de la Magdalena, 47005 Valladolid, Spain d Department of Cell and Molecular Biology, Box 596, Uppsala University, 751 24 Uppsala, Sweden e Department of Natural Sciences, Orebro¨ University, 701 82 Orebro,¨ Sweden Received 27 May 2004; accepted 14 September 2004 Available online 25 November 2004 Abstract The methods of the quantum electronic structure theory are reviewed and their implementation for the gas phase chemistry emphasized. Ab initio molecular orbital theory, density functional theory, quantum Monte Carlo theory and the methods to calculate the rate of complex chemical reactions in the gas phase are considered. Relativistic effects, other than the spin–orbit coupling effects, have not been considered. Rather than write down the main equations without further comments on how they were obtained, we provide the reader with essentials of the background on which the theory has been developed and the equations derived. We committed ourselves to place equations in their own proper perspective, so that the reader can appreciate more profoundly the subtleties of the theory underlying the equations themselves. Finally, a number of examples that illustrate the application of the theory are presented and discussed. -

Polar Covalent Bonds: Electronegativity
Polar Covalent Bonds: Electronegativity Covalent bonds can have ionic character These are polar covalent bonds Bonding electrons attracted more strongly by one atom than by the other Electron distribution between atoms is not symmetrical Bond Polarity and Electronegativity Symmetrical Covalent Bonds Polar Covalent Bonds C – C + - C – H C – O (non-polar) (polar) Electronegativity (EN): intrinsic ability of an atom to attract the shared electrons in a covalent bond Inductive Effect: shifting of sigma bonded electrons in resppygonse to nearby electronegative atom The Periodic Table and Electronegativity C – H C - Br and C - I (non-pol)lar) (po l)lar) Bond Polarity and Inductive Effect Nonpolar Covalent Bonds: atoms with similar EN Polar Covalent Bonds: Difference in EN of atoms < 2 Ionic Bonds: Difference in EN > 2 C–H bonds, relatively nonpolar C-O, C-X bonds (more electronegative elements) are polar Bonding electrons shift toward electronegative atom C acqqppuires partial positive char g,ge, + Electronegative atom acquires partial negative charge, - Inductive effect: shifting of electrons in a bond in response to EN of nearby atoms Electrostatic Potential Maps Electrostatic potential maps show calculated charge distributions Colors indicate electron- rich (red) and electron- poor (blue ) reg ions Arrows indicate direction of bond polarity Polar Covalent Bonds: Net Dipole Moments Molecules as a whole are often polar from vector summation of individual bond polarities and lone-pair contributions Strongly polar substances soluble in polar solvents like water; nonpolar substances are insoluble in water. Dipole moment ( ) - Net molecular polarity, due to difference in summed charges - magnitude of charge Q at end of molecular dipole times distance r between charges = Q r, in debyes (D), 1 D = 3.336 1030 coulomb meter length of an average covalent bond, the dipole moment would be 1.60 1029 Cm, or 4.80 D. -

Introduction to Molecular Orbital Theory
Chapter 2: Molecular Structure and Bonding Bonding Theories 1. VSEPR Theory 2. Valence Bond theory (with hybridization) 3. Molecular Orbital Theory ( with molecualr orbitals) To date, we have looked at three different theories of molecular boning. They are the VSEPR Theory (with Lewis Dot Structures), the Valence Bond theory (with hybridization) and Molecular Orbital Theory. A good theory should predict physical and chemical properties of the molecule such as shape, bond energy, bond length, and bond angles.Because arguments based on atomic orbitals focus on the bonds formed between valence electrons on an atom, they are often said to involve a valence-bond theory. The valence-bond model can't adequately explain the fact that some molecules contains two equivalent bonds with a bond order between that of a single bond and a double bond. The best it can do is suggest that these molecules are mixtures, or hybrids, of the two Lewis structures that can be written for these molecules. This problem, and many others, can be overcome by using a more sophisticated model of bonding based on molecular orbitals. Molecular orbital theory is more powerful than valence-bond theory because the orbitals reflect the geometry of the molecule to which they are applied. But this power carries a significant cost in terms of the ease with which the model can be visualized. One model does not describe all the properties of molecular bonds. Each model desribes a set of properties better than the others. The final test for any theory is experimental data. Introduction to Molecular Orbital Theory The Molecular Orbital Theory does a good job of predicting elctronic spectra and paramagnetism, when VSEPR and the V-B Theories don't. -

Heterocycles 2 Daniel Palleros
Heterocycles 2 Daniel Palleros Heterocycles 1. Structures 2. Aromaticity and Basicity 2.1 Pyrrole 2.2 Imidazole 2.3 Pyridine 2.4 Pyrimidine 2.5 Purine 3. Π-excessive and Π-deficient Heterocycles 4. Electrophilic Aromatic Substitution 5. Oxidation-Reduction 6. DNA and RNA Bases 7. Tautomers 8. H-bond Formation 9. Absorption of UV Radiation 10. Reactions and Mutations Heterocycles 3 Daniel Palleros Heterocycles Heterocycles are cyclic compounds in which one or more atoms of the ring are heteroatoms: O, N, S, P, etc. They are present in many biologically important molecules such as amino acids, nucleic acids and hormones. They are also indispensable components of pharmaceuticals and therapeutic drugs. Caffeine, sildenafil (the active ingredient in Viagra), acyclovir (an antiviral agent), clopidogrel (an antiplatelet agent) and nicotine, they all have heterocyclic systems. O CH3 N HN O O N O CH 3 N H3C N N HN N OH O S O H N N N 2 N O N N O CH3 N CH3 caffeine sildenafil acyclovir Cl S N CH3 N N H COOCH3 nicotine (S)-clopidogrel Here we will discuss the chemistry of this important group of compounds beginning with the simplest rings and continuing to more complex systems such as those present in nucleic acids. Heterocycles 4 Daniel Palleros 1. Structures Some of the most important heterocycles are shown below. Note that they have five or six-membered rings such as pyrrole and pyridine or polycyclic ring systems such as quinoline and purine. Imidazole, pyrimidine and purine play a very important role in the chemistry of nucleic acids and are highlighted. -

8.3 Bonding Theories >
8.3 Bonding Theories > Chapter 8 Covalent Bonding 8.1 Molecular Compounds 8.2 The Nature of Covalent Bonding 8.3 Bonding Theories 8.4 Polar Bonds and Molecules 1 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Molecular Orbitals How are atomic and molecular orbitals related? 2 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • The model you have been using for covalent bonding assumes the orbitals are those of the individual atoms. • There is a quantum mechanical model of bonding, however, that describes the electrons in molecules using orbitals that exist only for groupings of atoms. 3 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • When two atoms combine, this model assumes that their atomic orbitals overlap to produce molecular orbitals, or orbitals that apply to the entire molecule. 4 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Just as an atomic orbital belongs to a particular atom, a molecular orbital belongs to a molecule as a whole. • A molecular orbital that can be occupied by two electrons of a covalent bond is called a bonding orbital. 5 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Sigma Bonds When two atomic orbitals combine to form a molecular orbital that is symmetrical around the axis connecting two atomic nuclei, a sigma bond is formed. • Its symbol is the Greek letter sigma (σ). -

Chemical Bonding & Chemical Structure
Chemistry 201 – 2009 Chapter 1, Page 1 Chapter 1 – Chemical Bonding & Chemical Structure ings from inside your textbook because I normally ex- Getting Started pect you to read the entire chapter. 4. Finally, there will often be a Supplement that con- If you’ve downloaded this guide, it means you’re getting tains comments on material that I have found espe- serious about studying. So do you already have an idea cially tricky. Material that I expect you to memorize about how you’re going to study? will also be placed here. Maybe you thought you would read all of chapter 1 and then try the homework? That sounds good. Or maybe you Checklist thought you’d read a little bit, then do some problems from the book, and just keep switching back and forth? That When you have finished studying Chapter 1, you should be sounds really good. Or … maybe you thought you would able to:1 go through the chapter and make a list of all of the impor- tant technical terms in bold? That might be good too. 1. State the number of valence electrons on the following atoms: H, Li, Na, K, Mg, B, Al, C, Si, N, P, O, S, F, So what point am I trying to make here? Simply this – you Cl, Br, I should do whatever you think will work. Try something. Do something. Anything you do will help. 2. Draw and interpret Lewis structures Are some things better to do than others? Of course! But a. Use bond lengths to predict bond orders, and vice figuring out which study methods work well and which versa ones don’t will take time. -
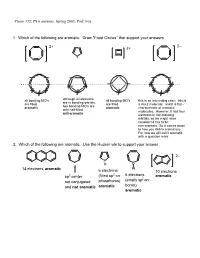
PS 6 Answers, Spring 2003, Prof
Chem 332, PS 6 answers, Spring 2003, Prof. Fox 1. Which of the following are aromatic. Draw "Frost Circles" that support your answers 2– 2+ 2+ + all bonding MO's although all electrons all bonding MO's this is an interesting case: this is are in bonding orbitals, are filled are filled a 4n+2 molecule, and it is flat-- two bonding MO's are aromatic aromatic characteristic of aromatic only half-filled molecules. However, it has four anti-aromatic electrons in non-bonding orbitals, so we might have considered this to be non-aromatic. So it comes down to how you define aromaticity. For now we will call it aromatic with a question mark. 2. Which of the following are aromatic. Use the Huckel rule to support your answer 2– P H B 14 electrons, aromatic H 6 electrons 10 electrons 2 6 electrons sp3 center (filled sp on aromatic (empty sp2 on not conjugated phosphorus) boron) and not aromatic aromatic aromatic 3. Compound 1 has an unusually large dipole moment for an organic hydrocarbon. Explain why (consider resonance structures for the central double bond) We need to remember that for any double bond compound, we can draw two polar resonance structure. Normally, these resonance structures are unimportant unimportant unimportant However, for 1 the story is different because there is a polar resonance form that has two aromatic components. In other words, 'breaking' the double bond gives rise to aromaticity, and therefore the polar resonance structure is important and leads to the observed dipole. dipole 1 8 e– 4 e– 6 e– 6 e– anti- anti- aromatic aromatic aromatic aromatic important resonance unimportant resonance structure structure 5. -

Covalent Bonding and Molecular Orbitals
Covalent Bonding and Molecular Orbitals Chemistry 35 Fall 2000 From Atoms to Molecules: The Covalent Bond n So, what happens to e- in atomic orbitals when two atoms approach and form a covalent bond? Mathematically: -let’s look at the formation of a hydrogen molecule: -we start with: 1 e-/each in 1s atomic orbitals -we’ll end up with: 2 e- in molecular obital(s) HOW? Make linear combinations of the 1s orbital wavefunctions: ymol = y1s(A) ± y1s(B) Then, solve via the SWE! 2 1 Hydrogen Wavefunctions wavefunctions probability densities 3 Hydrogen Molecular Orbitals anti-bonding bonding 4 2 Hydrogen MO Formation: Internuclear Separation n SWE solved with nuclei at a specific separation distance . How does the energy of the new MO vary with internuclear separation? movie 5 MO Theory: Homonuclear Diatomic Molecules n Let’s look at the s-bonding properties of some homonuclear diatomic molecules: Bond order = ½(bonding e- - anti-bonding e-) For H2: B.O. = 1 - 0 = 1 (single bond) For He2: B.O. = 1 - 1 = 0 (no bond) 6 3 Configurations and Bond Orders: 1st Period Diatomics Species Config. B.O. Energy Length + 1 H2 (s1s) ½ 255 kJ/mol 1.06 Å 2 H2 (s1s) 1 431 kJ/mol 0.74 Å + 2 * 1 He2 (s1s) (s 1s) ½ 251 kJ/mol 1.08 Å 2 * 2 He2 (s1s) (s 1s) 0 ~0 LARGE 7 Combining p-orbitals: s and p MO’s antibonding end-on overlap bonding antibonding side-on overlap bonding antibonding bonding8 4 2nd Period MO Energies s2p has lowest energy due to better overlap (end-on) of 2pz orbitals p2p orbitals are degenerate and at higher energy than the s2p 9 2nd Period MO Energies: Shift! For Z <8: 2s and 2p orbitals can interact enough to change energies of the resulting s2s and s2p MOs. -

Electron Configurations, Orbital Notation and Quantum Numbers
5 Electron Configurations, Orbital Notation and Quantum Numbers Electron Configurations, Orbital Notation and Quantum Numbers Understanding Electron Arrangement and Oxidation States Chemical properties depend on the number and arrangement of electrons in an atom. Usually, only the valence or outermost electrons are involved in chemical reactions. The electron cloud is compartmentalized. We model this compartmentalization through the use of electron configurations and orbital notations. The compartmentalization is as follows, energy levels have sublevels which have orbitals within them. We can use an apartment building as an analogy. The atom is the building, the floors of the apartment building are the energy levels, the apartments on a given floor are the orbitals and electrons reside inside the orbitals. There are two governing rules to consider when assigning electron configurations and orbital notations. Along with these rules, you must remember electrons are lazy and they hate each other, they will fill the lowest energy states first AND electrons repel each other since like charges repel. Rule 1: The Pauli Exclusion Principle In 1925, Wolfgang Pauli stated: No two electrons in an atom can have the same set of four quantum numbers. This means no atomic orbital can contain more than TWO electrons and the electrons must be of opposite spin if they are to form a pair within an orbital. Rule 2: Hunds Rule The most stable arrangement of electrons is one with the maximum number of unpaired electrons. It minimizes electron-electron repulsions and stabilizes the atom. Here is an analogy. In large families with several children, it is a luxury for each child to have their own room. -

Structure of Benzene, Orbital Picture, Resonance in Benzene, Aromatic Characters, Huckel’S Rule B
Dr.Mrityunjay Banerjee, IPT, Salipur B PHARM 3rd SEMESTER BP301T PHARMACEUTICAL ORGANIC CHEMISTRY –II UNIT I UNIT I Benzene and its derivatives 10 Hours A. Analytical, synthetic and other evidences in the derivation of structure of benzene, Orbital picture, resonance in benzene, aromatic characters, Huckel’s rule B. Reactions of benzene - nitration, sulphonation, halogenationreactivity, Friedelcrafts alkylation- reactivity, limitations, Friedelcrafts acylation. C. Substituents, effect of substituents on reactivity and orientation of mono substituted benzene compounds towards electrophilic substitution reaction D. Structure and uses of DDT, Saccharin, BHC and Chloramine Benzene and its Derivatives Chemists have found it useful to divide all organic compounds into two broad classes: aliphatic compounds and aromatic compounds. The original meanings of the words "aliphatic" (fatty) and "aromatic" (fragrant/ pleasant smell). Aromatic compounds are benzene and compounds that resemble benzene in chemical behavior. Aromatic properties are those properties of benzene that distinguish it from aliphatic hydrocarbons. Benzene: A liquid that smells like gasoline Boils at 80°C & Freezes at 5.5°C It was formerly used to decaffeinate coffee and component of many consumer products, such as paint strippers, rubber cements, and home dry-cleaning spot removers. A precursor in the production of plastics (such as Styrofoam and nylon), drugs, detergents, synthetic rubber, pesticides, and dyes. It is used as a solvent in cleaning and maintaining printing equipment and for adhesives such as those used to attach soles to shoes. Benzene is a natural constituent of petroleum products, but because it is a known carcinogen, its use as an additive in gasoline is now limited. In 1970s it was associated with leukemia deaths.