Mechanistic Studies on Aldehyde Decarbonylase from Cyanobacteria: a New Enzyme for Alkane Biosynthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Dynamics Simulation Studies of Physico of Liquid
MD Simulation of Liquid Pentane Isomers Bull. Korean Chem. Soc. 1999, Vol. 20, No. 8 897 Molecular Dynamics Simulation Studies of Physico Chemical Properties of Liquid Pentane Isomers Seng Kue Lee and Song Hi Lee* Department of Chemistry, Kyungsung University, Pusan 608-736, Korea Received January 15, 1999 We have presented the thermodynamic, structural and dynamic properties of liquid pentane isomers - normal pentane, isopentane, and neopentane - using an expanded collapsed atomic model. The thermodynamic prop erties show that the intermolecular interactions become weaker as the molecular shape becomes more nearly spherical and the surface area decreases with branching. The structural properties are well predicted from the site-site radial, the average end-to-end distance, and the root-mean-squared radius of gyration distribution func tions. The dynamic properties are obtained from the time correlation functions - the mean square displacement (MSD), the velocity auto-correlation (VAC), the cosine (CAC), the stress (SAC), the pressure (PAC), and the heat flux auto-correlation (HFAC) functions - of liquid pentane isomers. Two self-diffusion coefficients of liq uid pentane isomers calculated from the MSD's via the Einstein equation and the VAC's via the Green-Kubo relation show the same trend but do not coincide with the branching effect on self-diffusion. The rotational re laxation time of liquid pentane isomers obtained from the CAC's decreases monotonously as branching increas es. Two kinds of viscosities of liquid pentane isomers calculated from the SAC and PAC functions via the Green-Kubo relation have the same trend compared with the experimental results. The thermal conductivity calculated from the HFAC increases as branching increases. -

Particulate Preparation from Pisum Sativum (Plant Wax/CO Production/Hydrocarbon) T
Proc. Natl. Acad. Sci. USA Vol. 81, pp. 6613-6617, November 1984 Biochemistry Alkane biosynthesis by decarbonylation of aldehydes catalyzed by a particulate preparation from Pisum sativum (plant wax/CO production/hydrocarbon) T. M. CHEESBROUGH AND P. E. KOLATTUKUDY* Institute of Biological Chemistry and Biochemistry/Biophysics Program, Washington State University. Pullman, WA 99164-6340 Communicated by P. K. Stumpf, June 29, 1984 ABSTRACT Mechanism of enzymatic conversion of a fat- MATERIALS AND METHODS ty acid to the corresponding alkane by the loss of the carboxyl Materials. Omnifluor, [1-_4C]stearic acid, [U-3H]tetraco- carbon was investigated with particulate preparations from Pi- sanoic acid, [1-'4Cjsteroyl-CoA, and LiAl3H4 were from sum sativum. A heavy particulate preparation (sp. gr., 1.30 New England Nuclear. Sodium [1-14C]cyanide was from g/cm3) isolated by two density-gradient centrifugation steps ICN (Chemical and Radioisotopes Division). Pyridinium catalyzed conversion of octadecanal to heptadecane and CO. chlorochromate, meso-tetraphenylporphyrin, and RhCP Experiments with [1-3H,1_14C]octadecanal showed the stoichi- 3H2O were from Aldrich. LiAIH4, di-t-butylchlorophos- ometry of the reaction and retention of the aldehydic hydrogen phine, and Ru(CO)12 were from Alfa Products. The rhodium in the alkane during this enzymatic decarbonylation. This de- chelate was synthesized by the procedure of Monson (8). carbonylase showed an optimal pH of 7.0 and a Km of 35 ,uM Ruthenium dicarbonyl tetraphenylporphyrin [Ru(CO)2- for the aldehyde. This enzyme was severely inhibited by metal (TTP)] was synthesized (9) and activated by substitution of ion chelators and showed no requirement for any cofactors. -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Supplementary Material (ESI) for Chemical Communications This Journal Is © the Royal Society of Chemistry 2012
Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Supplementary Material Guest-controlled Self-Sorting in Assemblies driven by the Hydrophobic Effect Haiying Gan, Bruce C. Gibb* Department of Chemistry University of New Orleans LA 70148 Email: [email protected] S1 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Table of Contents General p S3 1H-NMR spectra of 1 and 2, and mixture of hosts, in the absence of guests p S4 1H-NMR spectra of the complexes of 1.2 and hydrocarbons n-pentane through n-heptadecane p S5-S8 Example COSY and NOESY NMR data (for n-undecane hetero-complex) p S9 Pulse-gradient stimulated spin-echo NMR studies p S11 Table of Percentage of Hetero-complex Formation p S12 References p S13 S2 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Experimental section General All reagents were purchased from Aldrich Chemical Company and used as received without further purification. Host 1 and 2 were synthesized as previously reported.1,2 NMR spectra were recorded on a Varian Inova 500 MHz spectrometer. Chemical shifts are reported relative to D2O (4.80 ppm). Endo Me c O O O O O O O O O O g O O g O O O d O d HO O HO O HO f O OH OH HO f O OH OH O b O O b O H H H H H H H H O e O e O O O O O O O O O O O O O O j j H H a H H H Ha H H m l m l HO O HO O O OH O OH HO O HO O O OH O OH 1 2 S3 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 1H-NMR spectra of host 1, 2 and mixture of hosts in the absence of guests Figure S1: 1H NMR spectra of: 1) 1 mM of 1; 2) 1 mM of 2; 3) 0.5 mM of both 1 and 2 (all 10 mM sodium tetraborate solution). -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
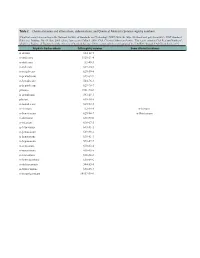
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Synthesis and Reactivity of Cyclopentadienyl Based Organometallic Compounds and Their Electrochemical and Biological Properties
Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Sasmita Mishra Department of Chemistry National Institute of Technology Rourkela Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Dissertation submitted to the National Institute of Technology Rourkela In partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry by Sasmita Mishra (Roll Number: 511CY604) Under the supervision of Prof. Saurav Chatterjee February, 2017 Department of Chemistry National Institute of Technology Rourkela Department of Chemistry National Institute of Technology Rourkela Certificate of Examination Roll Number: 511CY604 Name: Sasmita Mishra Title of Dissertation: ''Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties We the below signed, after checking the dissertation mentioned above and the official record book(s) of the student, hereby state our approval of the dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry at National Institute of Technology Rourkela. We are satisfied with the volume, quality, correctness, and originality of the work. --------------------------- Prof. Saurav Chatterjee Principal Supervisor --------------------------- --------------------------- Prof. A. Sahoo. Prof. G. Hota Member (DSC) Member (DSC) --------------------------- -

LE Hatch, W. Luo, JF Pankow, RJ Yokelson 2, CE Stockwell 2, and KC Barsanti
Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Atmos. Chem. Phys. Discuss., 14, 23237–23307, 2014 www.atmos-chem-phys-discuss.net/14/23237/2014/ doi:10.5194/acpd-14-23237-2014 ACPD © Author(s) 2014. CC Attribution 3.0 License. 14, 23237–23307, 2014 This discussion paper is/has been under review for the journal Atmospheric Chemistry Identification of and Physics (ACP). Please refer to the corresponding final paper in ACP if available. NMOCs in biomass burning smoke Identification and quantification of L. E. Hatch et al. gaseous organic compounds emitted from biomass burning using Title Page Abstract Introduction two-dimensional gas Conclusions References chromatography/time-of-flight mass Tables Figures spectrometry J I L. E. Hatch1, W. Luo1, J. F. Pankow1, R. J. Yokelson2, C. E. Stockwell2, and J I 1 K. C. Barsanti Back Close 1 Department of Civil and Environmental Engineering, Portland State University, Portland, Full Screen / Esc Oregon 2 Department of Chemistry, University of Montana, Missoula, Montana Printer-friendly Version Received: 15 August 2014 – Accepted: 19 August 2014 – Published: 10 September 2014 Interactive Discussion Correspondence to: K. C. Barsanti ([email protected]) Published by Copernicus Publications on behalf of the European Geosciences Union. 23237 Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Abstract ACPD The current understanding of secondary organic aerosol (SOA) formation within biomass burning (BB) plumes is limited by the incomplete identification and quan- 14, 23237–23307, 2014 tification of the non-methane organic compounds (NMOCs) emitted from such fires. 5 Gaseous organic compounds were collected on sorbent cartridges during labora- Identification of tory burns as part of the fourth Fire Lab at Missoula Experiment (FLAME-4), with NMOCs in biomass analysis by two-dimensional gas chromatography/time-of-flight mass spectrometry burning smoke (GC × GC/TOFMS). -

N-Alkane Category: Decane, Undecane, Dodecane (CAS Nos
June 17, 2004 n-Alkane Category: decane, undecane, dodecane (CAS Nos. 124-18-5, 1120-21-4, 112-40-3) Voluntary Children’s Chemical Evaluation Program (VCCEP) Tier 1 Pilot Submission Docket Number OPPTS – 00274D American Chemistry Council n-Alkane VCCEP Consortium Sponsors: Chevron Phillips Chemical Company LP Sasol North America Inc. Shell Chemical LP June 17, 2004 TABLE OF CONTENTS Glossary of Terms 4 1. Executive Summary 5 2. Basis for Inclusion in the VCCEP Program 2.1 Total Exposure Assessment Methodology Data 10 2.2 Air Monitoring Data 11 2.3 How Sponsors Were Identified for the n-Alkane VCCEP Effort 12 3. Previous and On-Going Health Assessments 3.1 OECD SIDS/ICCA HPV Imitative 14 3.2 Total Petroleum Hydrocarbon Criteria Working Group 15 3.3 Hydrocarbon Solvent Guidance Group Values (GGVs) for Setting Occupational Exposure Limits (OELs) for Hydrocarbon Solvents 15 4. Regulatory Overview 4.1 CPSC Child-Resistant Packaging for Hydrocarbons 16 4.2 Occupational Exposure Limits 16 4.3 VOC Regulations 17 5. Product Overview 5.1 Physical, Chemical, and Environmental Fate Properties 18 5.2 Production of n-Alkanes 20 5.3 Uses for n-Alkane Products 20 5.4 Petroleum Products Which Contain n-Alkanes 21 6. Exposure Assessment 6.1 Summary 25 6.2 Non-Occupational Exposure 28 6.2.1 Indoor Sources of Exposure 6.2.2 Outdoor Source of Exposure 6.2.3 Unique Children’s Exposure 6.3 Integrated 24 hour Exposure 36 6.4 Occupational Exposure 36 6.5 Potential for Dermal and Oral Exposure 40 6.6 Selection of Exposure Scenarios and Exposure Concentrations 43 7. -

Fusing Catalase to an Alkane-Producing Enzyme
Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2 Carl Andre1, Sung Won Kim, Xiao-Hong Yu, and John Shanklin2 Biosciences Department, Brookhaven National Laboratory, Upton, NY 11973 Edited by Rodney B. Croteau, Washington State University, Pullman, WA, and approved December 31, 2012 (received for review October 29, 2012) Biologically produced alkanes represent potential renewable alter- enzyme was determined as part of a structural genomics effort natives to petroleum-derived chemicals. A cyanobacterial pathway (Protein Data Bank 2OC5, Joint Center for Structural Genom- consisting of acyl–Acyl Carrier Protein reductase and an aldehyde- ics). ADO converts aldehydes to n-1 alkanes with the aldehyde C1 deformylating oxygenase (ADO) converts acyl–Acyl Carrier Pro- released as formic acid (8, 9). Electrons required for the reaction teins into corresponding n-1 alkanes via aldehyde intermediates can be provided by NADPH via ferredoxin-NADP reductase in an oxygen-dependent manner (Km for O2,84± 9 μM). In vitro, (FNR), and ferredoxin (Fd) (7), or via the chemical mediator ADO turned over only three times, but addition of more ADO to phenazine methosulfate (PMS) (10). A proposed reaction scheme exhausted assays resulted in additional product formation. While for ADO (adapted from ref. 9) is depicted below where R evaluating the peroxide shunt to drive ADO catalysis, we discov- represents Acyl: ered that ADO is inhibited by hydrogen peroxide (H2O2) with an apparent Ki of 16 ± 6 μM and that H2O2 inhibition is of mixed-type with respect to O2. Supplementing exhausted assays with catalase (CAT) restored ADO activity, demonstrating that inhibition was reversible and dependent on H2O2, which originated from poor coupling of reductant consumption with alkane formation. -

Pomona Oaks Exposure Assessment
POMONA OAKS EXPOSURE ASSESSMENT Volatile Organics in Well Water and Indoor Air Environmental Health Program Division of Epidemiology and Disease Control New Jersey State Department of Health October TABLE OF CONTENTS List of Illustrations List of Tables Acknowiedgem ents SECTION 1. INTRODUCTION 1.1. Volatile Organics in the Community 1.2. Benzene - A Contaminant of Particular Concern 1.3. Initiation of Investigation 1.*. Study Objectives 1.5. Description of Population Studied SECTION 2. MATERIALS AND METHODS 2.1. Pilot Study 2.1.a. Study Design 2.1.b. Sample Selection and Participation 2.I.e. Data Collection and Laboratory Analysis 2.1.d. Data Management and Statistical Methods 2.2. Full Scale Study 2.2.a. Study Design 2.2.b.. Sample Selection and Participation 2.5.C. Data Collection and Laboratory Analysis 2.2.d. Data Management and Statistical Methods 2.3 Follow-up Study 2.3.a. Study Design 2.3.b. Sample Selection and Participation 2.3.C. Data Collection and Laboratory Analysis 2.3.d. Data Management and Statistical Methods 2A Methods of Communication SECTION 3. RESULTS 3.1. Pilot Study Results 3.1.a. Sample Studied 3.1.b. Exposures 3.1.c. Response 3.1.d. Exposure-Response Relationship TABLE OF CONTENTS (CONTINUED) 3.2. Full Scale Study Results 3.2.a. Sample Studied 3.2.b. Exposures 3.2.C. Response 3.2.d. Exposure-Response Relationship 3.3. Follow-up Study Results 3.3.a. Sample Studied 3.3.b. Exposures 3.3.C. Risk Assessment SECTION 4. DISCUSSION 4.1. Pilot Study 4.2.