Histone-Binding of DPF2 Mediates Its Repressive Role in Myeloid Differentiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ACTL6A Promotes the Proliferation of Esophageal Squamous Cell Carcinoma Cells and Correlates with Poor Clinical Outcomes
OncoTargets and Therapy Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH ACTL6A Promotes the Proliferation of Esophageal Squamous Cell Carcinoma Cells and Correlates with Poor Clinical Outcomes This article was published in the following Dove Press journal: OncoTargets and Therapy Rui-zhe Li1 Background: ACTL6A, a regulatory subunit of ATP-dependent chromatin-remodeling Yun-yun Li1,2 complexes SWI/SNF, has been identified as a central oncogenic driver in many tumor types. Hui Qin1 Materials and Methods: We used immunohistochemistry (IHC) to detect ACTL6A Shan-shan Li1 expression in esophageal squamous cell carcinoma (ESCC) tissues. Then, the effect of ACTL6A on proliferation and DNA synthesis was explored by using cell counting kit 8 1 Department of Pathology, School of (CCK8) and EdU retention assays. The potential oncogenic mechanism of ACTL6A in Basic Medical Sciences, Zhengzhou University and First Affiliated Hospital of ESCC cells was also analyzed by flow cytometry and Western blotting. We further estab Zhengzhou University, Zhengzhou, lished an ESCC xenograft mouse model to validate the in vitro results. Henan 450000, People’s Republic of China; 2Department of Stomatology, First Results: ACTL6A expression, localized in cancer cell nuclei, was markedly higher in ESCC Affiliated Hospital of Zhengzhou tissues than in the corresponding noncancerous tissues (P<0.001) and was positively asso University, Zhengzhou, Henan 450000, ciated with tumor size, histological differentiation, T stage and tumor-node-metastasis People’s Republic of China (TNM) stage. Kaplan–Meier analysis revealed that high ACTL6A expression was signifi cantly associated with poor overall survival (OS) (P = 0.008, HR= 2.562, 95% CI: 1.241– 5.289), and decision curve analysis (DCA) demonstrated that ACTL6A could increase the clinical prognostic efficiency of the original clinical prediction model. -

Integrase Interactor 1 (INI-1) Deficient Renal Cell Carcinoma
Open Access Case Report DOI: 10.7759/cureus.13082 Integrase Interactor 1 (INI-1) Deficient Renal Cell Carcinoma Manpreet Singh 1 , Harkirat Singh 1 , Benjamin Hambro 1 , Jasleen Kaur 1 , Ravi Rao 2 1. Internal Medicine, St Agnes Medical Center, Fresno, USA 2. Hematology and Oncology, St Agnes Medical Center, Fresno, USA Corresponding author: Manpreet Singh, [email protected] Abstract Members of the SWItch/sucrose nonfermentable (SWI-SNF) family, including SWI/SNF related, matrix- associated, actin-dependent regulator of chromatin, subfamily A, member 4 (SMARCA4), SWI/SNF related, matrix‐associated, actin‐dependent regulator of chromatin, subfamily B member 1 (SMARCB1)/integrase interactor 1 (INI-1) are known tumor suppressor genes. Interactions between SMARCB1/INI-1 and key protein components in various cellular pathways are related to tumor progression and proliferation. SMARCB1/INI-1 protein was undetectable in rhabdoid tumor cells, whereas non-tumorous cells express the SMARCB1/INI-1 genes. Germline and sporadic mutations of several genes encoding for proteins in this complex are known to cause a spectrum of cancers, usually with sarcomatoid features which include a very aggressive renal medullary carcinoma. We report a case of a 29-year-old male who presented with SMARCA4 deficient renal tumor with a very aggressive clinical behavior which ultimately led to his death. Categories: Nephrology, Oncology Keywords: renal medullary carcinoma, ini-1, renal cell carcinoma, swi/snf, smarcb1, integrase interactor 1 Introduction Renal medullary carcinoma is a rare and very aggressive malignancy affecting young adults with rare cases in patients with sickle cell disease or trait [1]. The tumor arises predominantly in the renal medulla and exhibits a variety of growth patterns including reticular, solid, tubular, trabecular, cribriform, sarcomatoid, and micropapillary [1]. -

Bioinformatics Applications Through Visualization of Variations on Protein
1 Bioinformatics applications through visualization of variations on protein structures, comparative functional genomics, and comparative modeling for protein structure studies A dissertation presented by Alper Uzun to The Department of Biology In partial fulfillment of the requirements for the degree of Doctor of Philosophy in the field of Biology Northeastern University Boston, Massachusetts July 2009 2 ©2009 Alper Uzun ALL RIGHTS RESERVED 3 Bioinformatics applications through visualization of variations on protein structures, comparative functional genomics, and comparative modeling for protein structure studies by Alper Uzun ABSTRACT OF DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology in the Graduate School of Arts and Sciences of Northeastern University, July, 2009 4 Abstract The three-dimensional structure of a protein provides important information for understanding and answering many biological questions in molecular detail. The rapidly growing number of sequenced genes and related genomic information is intensively accumulating in the biological databases. It is significantly important to combine biological data and developing bioinformatics tools while information of protein sequences, structures and DNA sequences are exponentially growing. On the other hand, especially the number of known protein sequences is much larger than the number of experimentally solved protein structures. However the experimental methods cannot always be applied or protein structures -

Increased ACTL6A Occupancy Within Mswi/SNF Chromatin Remodelers
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.22.435873; this version posted March 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Increased ACTL6A Occupancy Within mSWI/SNF Chromatin Remodelers Drives Human Squamous Cell Carcinoma Chiung-Ying Chang1,2,7, Zohar Shipony3,7, Ann Kuo1,2, Kyle M. Loh4,5, William J. Greenleaf3,6, Gerald R. Crabtree1,2,5* 1Howard Hughes Medical Institute, Stanford University School of Medicine, Stanford, California 94305, USA. 2Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, USA. 3Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA. 4Institute for Stem Cell Biology and Regenerative Medicine, Stanford University School of Medicine, Stanford, California 94305, USA. 5Department of Developmental Biology, Stanford University School of Medicine, Stanford, California 94305, USA. 6Department of Applied Physics, Stanford University, Stanford, California 94305, USA. 7These authors contributed equally. *Corresponding author: Gerald R. Crabtree, [email protected] Summary Mammalian SWI/SNF (BAF) chromatin remodelers play dosage-sensitive roles in many human malignancies and neurologic disorders. The gene encoding the BAF subunit, ACTL6A, is amplified at an early stage in the development of squamous cell carcinomas (SCCs), but its oncogenic role remains unclear. Here we demonstrate that ACTL6A overexpression leads to its stoichiometric assembly into BAF complexes and drives its interaction and engagement with specific regulatory regions in the genome. In normal epithelial cells, ACTL6A was sub-stoichiometric to other BAF subunits. However, increased ACTL6A levels by ectopic expression or in SCC cells led to near-saturation of ACTL6A within BAF complexes. -

Loss of Integrase Interactor 1 (INI1) Expression in a Subset of Differentiated Thyroid Cancer
diagnostics Article Loss of Integrase Interactor 1 (INI1) Expression in a Subset of Differentiated Thyroid Cancer Kung-Chen Ho 1, Jie-Jen Lee 1, Chi-Hsin Lin 2,3, Ching-Hsiang Leung 4 and Shih-Ping Cheng 1,5,* 1 Department of Surgery, MacKay Memorial Hospital and Mackay Medical College, Taipei 104215, Taiwan; [email protected] (K.-C.H.); [email protected] (J.-J.L.) 2 Department of Medical Research, MacKay Memorial Hospital, Taipei 104215, Taiwan; [email protected] 3 Department of Bioscience Technology, Chung Yuan Christian University, Taoyuan City 320314, Taiwan 4 Division of Endocrinology and Metabolism, Department of Internal Medicine, MacKay Memorial Hospital and Mackay Medical College, Taipei 104215, Taiwan; [email protected] 5 Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University, Taipei 110301, Taiwan * Correspondence: [email protected]; Tel.: +886-2-2543-3535 Received: 19 April 2020; Accepted: 2 May 2020; Published: 5 May 2020 Abstract: Alterations in the switching defective/sucrose non-fermenting (SWI/SNF) chromatin-remodeling complex are enriched in advanced thyroid cancer. Integrase interactor 1 (INI1), encoded by the SMARCB1 gene on the long arm of chromosome 22, is one of the core subunits of the SWI/SNF complex. INI1 immunohistochemistry is frequently used for the diagnosis of malignant rhabdoid neoplasms. In the present study, we found normal and benign thyroid tissues generally had diffusely intense nuclear immunostaining. Loss of INI1 immunohistochemical expression was observed in 8% of papillary thyroid cancer and 30% of follicular thyroid cancer. Furthermore, loss of INI1 expression was associated with extrathyroidal extension (p < 0.001) and lymph node metastasis (p = 0.038). -

Renal Medullary Carcinomas Depend Upon SMARCB1 Loss And
RESEARCH ARTICLE Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition Andrew L Hong1,2,3, Yuen-Yi Tseng3, Jeremiah A Wala3, Won-Jun Kim2, Bryan D Kynnap2, Mihir B Doshi3, Guillaume Kugener3, Gabriel J Sandoval2,3, Thomas P Howard2, Ji Li2, Xiaoping Yang3, Michelle Tillgren2, Mahmhoud Ghandi3, Abeer Sayeed3, Rebecca Deasy3, Abigail Ward1,2, Brian McSteen4, Katherine M Labella2, Paula Keskula3, Adam Tracy3, Cora Connor5, Catherine M Clinton1,2, Alanna J Church1, Brian D Crompton1,2,3, Katherine A Janeway1,2, Barbara Van Hare4, David Sandak4, Ole Gjoerup2, Pratiti Bandopadhayay1,2,3, Paul A Clemons3, Stuart L Schreiber3, David E Root3, Prafulla C Gokhale2, Susan N Chi1,2, Elizabeth A Mullen1,2, Charles WM Roberts6, Cigall Kadoch2,3, Rameen Beroukhim2,3,7, Keith L Ligon2,3,7, Jesse S Boehm3, William C Hahn2,3,7* 1Boston Children’s Hospital, Boston, United States; 2Dana-Farber Cancer Institute, Boston, United States; 3Broad Institute of Harvard and MIT, Cambridge, United States; 4Rare Cancer Research Foundation, Durham, United States; 5RMC Support, North Charleston, United States; 6St. Jude Children’s Research Hospital, Memphis, United States; 7Brigham and Women’s Hospital, Boston, United States Abstract Renal medullary carcinoma (RMC) is a rare and deadly kidney cancer in patients of African descent with sickle cell trait. We have developed faithful patient-derived RMC models and using whole-genome sequencing, we identified loss-of-function intronic fusion events in one SMARCB1 allele with concurrent loss of the other allele. Biochemical and functional characterization of these models revealed that RMC requires the loss of SMARCB1 for survival. -

Identification of a Core Member of the SWI/SNF Complex, BAF155/SMARCC1, As a Human Tumor Suppressor Gene
RESEARCH PAPER Epigenetics 6:12, 1444-1453; December 2011; © 2011 Landes Bioscience Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene Jessica DelBove,1,6 Gary Rosson,2,7 Matthew Strobeck,3 Jianguang Chen,4 Trevor K. Archer,4 Weidong Wang,5 Erik S. Knudsen3 and Bernard E. Weissman1,2,* 1Department of Pathology and Laboratory Medicine; 2University of North Carolina-Lineberger Comprehensive Cancer Center; 7Department of Genetics; University of North Carolina; Chapel Hill, NC USA; 3Department of Cancer Biology and the Kimmel Cancer Center; Thomas Jefferson University; Philadelphia, PA USA; 4Chromatin and Gene Expression Section; Laboratory of Molecular Carcinogenesis; National Institute of Environmental Health Sciences; National Institutes of Health; Research Triangle Park, NC USA; 5Laboratory of Genetics; National Institute on Aging; National Institutes of Health; Baltimore, MD USA; 6Department of Hematology; University of Utah School of Medicine; Salt Lake City, UT USA Key words: SWI/SNF, BAF155, SMARCC1, tumor suppressor gene, cancer epigenetics Recent studies have established that two core members of the SWI/SNF chromatin remodeling complex, BRG1 and SNF5/ INI1, possess tumor-suppressor activity in human and mouse cancers. While the third core member, BAF155, has been implicated by several studies as having a potential role in tumor development, direct evidence for its tumor suppressor activity has remained lacking. Therefore, we screened for BAF155 deficiency in a large number of human tumor cell lines. We identified two cell lines, the SNUC2B colon carcinoma and the SKOV3 ovarian carcinoma, displaying a complete loss of protein expression while maintaining normal levels of mRNA expression. -

Expressed Gene Fusions As Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer
Published OnlineFirst December 14, 2017; DOI: 10.1158/2159-8290.CD-17-0535 RESEARCH ARTICLE Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer Karina J. Matissek1,2, Maristela L. Onozato3, Sheng Sun1,2, Zongli Zheng2,3,4, Andrew Schultz1, Jesse Lee3, Kristofer Patel1, Piiha-Lotta Jerevall2,3, Srinivas Vinod Saladi1,2, Allison Macleay3, Mehrad Tavallai1,2, Tanja Badovinac-Crnjevic5, Carlos Barrios6, Nuran Beşe7, Arlene Chan8, Yanin Chavarri-Guerra9, Marcio Debiasi6, Elif Demirdögen10, Ünal Egeli10, Sahsuvar Gökgöz10, Henry Gomez11, Pedro Liedke6, Ismet Tasdelen10, Sahsine Tolunay10, Gustavo Werutsky6, Jessica St. Louis1, Nora Horick12, Dianne M. Finkelstein2,12, Long Phi Le2,3, Aditya Bardia1,2, Paul E. Goss1,2, Dennis C. Sgroi2,3, A. John Iafrate2,3, and Leif W. Ellisen1,2 ABSTRACT We sought to uncover genetic drivers of hormone receptor–positive (HR+) breast cancer, using a targeted next-generation sequencing approach for detecting expressed gene rearrangements without prior knowledge of the fusion partners. We identified inter- genic fusions involving driver genes, including PIK3CA, AKT3, RAF1, and ESR1, in 14% (24/173) of unselected patients with advanced HR+ breast cancer. FISH confirmed the corresponding chromo- somal rearrangements in both primary and metastatic tumors. Expression of novel kinase fusions in nontransformed cells deregulates phosphoprotein signaling, cell proliferation, and survival in three- dimensional culture, whereas expression in HR+ breast cancer models modulates estrogen-dependent growth and confers hormonal therapy resistance in vitro and in vivo. Strikingly, shorter overall survival was observed in patients with rearrangement-positive versus rearrangement-negative tumors. Cor- respondingly, fusions were uncommon (<5%) among 300 patients presenting with primary HR+ breast cancer. -

Inactivation of the PBRM1 Tumor Suppressor Gene Amplifies
Inactivation of the PBRM1 tumor suppressor gene − − amplifies the HIF-response in VHL / clear cell renal carcinoma Wenhua Gaoa, Wei Lib,c, Tengfei Xiaoa,b,c, Xiaole Shirley Liub,c, and William G. Kaelin Jr.a,d,1 aDepartment of Medical Oncology, Dana-Farber Cancer Institute and Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; bCenter for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, MA 02215; cDepartment of Biostatistics and Computational Biology, Dana-Farber Cancer Institute and Harvard T.H. Chan School of Public Health, Boston, MA 02115; and dHoward Hughes Medical Institute, Chevy Chase, MD 20815 Contributed by William G. Kaelin, Jr., December 1, 2016 (sent for review October 31, 2016; reviewed by Charles W. M. Roberts and Ali Shilatifard) Most clear cell renal carcinomas (ccRCCs) are initiated by somatic monolayer culture and in soft agar (10). These effects were not, inactivation of the VHL tumor suppressor gene. The VHL gene prod- however, proven to be on-target, and were not interrogated in uct, pVHL, is the substrate recognition unit of an ubiquitin ligase vivo. As a step toward understanding the role of BAF180 in that targets the HIF transcription factor for proteasomal degrada- ccRCC, we asked whether BAF180 participates in the canonical tion; inappropriate expression of HIF target genes drives renal car- PBAF complex in ccRCC cell lines and whether loss of BAF180 cinogenesis. Loss of pVHL is not sufficient, however, to cause ccRCC. measurably alters ccRCC behavior in cell culture and in mice. Additional cooperating genetic events, including intragenic muta- tions and copy number alterations, are required. -

Rats and Axolotls Share a Common Molecular Signature After Spinal Cord Injury Enriched in Collagen-1
Rats and axolotls share a common molecular signature after spinal cord injury enriched in collagen-1 Athanasios Didangelos1, Katalin Bartus1, Jure Tica1, Bernd Roschitzki2, Elizabeth J. Bradbury1 1Wolfson CARD King’s College London, United Kingdom. 2Centre for functional Genomics, ETH Zurich, Switzerland. Running title: spinal cord injury in rats and axolotls Correspondence: A Didangelos: [email protected] SUPPLEMENTAL FIGURES AND LEGENDS Supplemental Fig. 1: Rat 7 days microarray differentially regulated transcripts. A-B: Protein-protein interaction networks of upregulated (A) and downregulated (B) transcripts identified by microarray gene expression profiling of rat SCI (4 sham versus 4 injured spinal cord samples) 7 days post-injury. Microarray expression data and experimental information is publicly available online (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45006) and is also summarised in Supplemental Table 1. Protein-protein interaction networks were performed in StringDB using the full range of protein interaction scores (0.15 – 0.99) to capture maximum evidence of proteins’ interactions. Networks were then further analysed for betweeness centrality and gene ontology (GO) annotations (BinGO) in Cytoscape. Node colour indicates betweeness centrality while edge colour and thickness indicate interaction score based on predicted functional links between nodes (green: low values; red: high values). C-D: The top 10 upregulated (C) or downregulated (D) transcripts sorted by betweeness centrality score in protein-protein interaction networks shown in A & B. E-F: Biological process GO analysis was performed on networks of upregulated and downregulated genes using BinGO in Cytoscape. Graphs indicate the 20 most significant GO categories and the number of genes in each category. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -
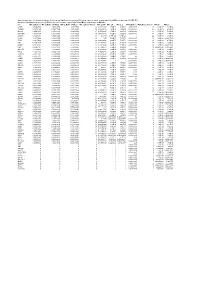
Supplementary Table 6. Summary of Mutation Frequency And
Supplementary table 6. Summary of mutation frequency and significance per microsatellite and per gene in exome sequencing of 24 and MiSeq sequencing of 93 MSI CRCs Summary of mutation frequency and significance per gene in exome sequencing of 24 and MiSeq sequencing of 93 MSI CRCs Gene MiSeq_MutFreq MiSeq_MutFreqCI95High MiSeq_MutFreqCI95Low MiSeq_MutatedTumors MiSeq_NAF MiSeq_P MiSeq_Q WXS_MutFreq WXS_MutatedTumors WXS_P WXS_Q CASP5 0.989247312 1.006838564 0.97165606 92 0.666666667 1.70E-89 4.83E-88 0.791666667 19 2.12E-17 1.33E-14 TGFBR2 0.967741935 0.997877874 0.937605997 90 0.883484163 1.48E-96 6.99E-95 0.833333333 20 4.39E-19 4.13E-16 SLC9A8 0.935483871 0.977386193 0.893581549 87 0.655034325 1.70E-89 4.83E-88 0.458333333 11 5.71E-07 5.25E-05 MIS18BP1 0.913978495 0.961803749 0.86615324 85 0.488888889 7.02E-61 9.07E-60 0.708333333 17 1.51E-08 1.97E-06 ACVR2A 0.892473118 0.945310572 0.839635665 83 0.80625 4.77E-128 6.77E-126 1 24 5.63E-36 2.12E-32 CDH26 0.88172043 0.936801974 0.826638886 82 0.55 4.12E-79 7.31E-78 0.416666667 10 5.46E-06 0.000331959 TFAM 0.88172043 0.936801974 0.826638886 82 0.620192308 4.12E-79 7.31E-78 0.583333333 14 2.52E-10 4.75E-08 ATR 0.860215054 0.919360254 0.801069854 80 0.632996633 2.72E-75 4.30E-74 0.583333333 14 3.89E-09 5.86E-07 AASDH 0.838709677 0.901442635 0.77597672 78 0.832712022 1.27E-71 1.80E-70 0.416666667 10 5.46E-06 0.000331959 USF3 0.806451613 0.873837619 0.739065607 75 0.771428571 5.39E-50 4.25E-49 0.5 12 0.002548628 0.01890901 SLC35F5 0.76344086 0.83592506 0.69095666 71 0.607908352 8.90E-60 1.05E-58