(12) Patent Application Publication (10) Pub. No.: US 2015/0337042 A1 Reilly Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 8,148,546 B2 Schuster Et Al
US008148546B2 (12) United States Patent (10) Patent No.: US 8,148,546 B2 Schuster et al. (45) Date of Patent: Apr. 3, 2012 (54) TETRAHYDROCARBAZOLE DERIVATIVES (58) Field of Classification Search .................. 548/448: ASLGANDS OF G-PROTEIN COUPLED 51474 11 RECEPTORS See application file for complete search history. (75) Inventors: Tilmann Schuster, Grossostheim (DE); Klaus Paulini, Maintal (DE); Peter (56) References Cited Schmidt, Schoeneck (DE); Silke Baasner, Schoeneck (DE); Emmanuel FOREIGN PATENT DOCUMENTS Polymeropoulos, Frankfurt (DE); WO WO O3051837 * 6, 2003 Eckhard Guenther, Maintal (DE); WO WO 2006005484 * 1, 2006 Michael Teifel, Weiterstadt (DE) OTHER PUBLICATIONS (73) Assignee: AEterna Zentaris GmbH, Frankfurt Kubinyi (3D QSAR in Drug Design: Ligand-Protein Interactions and (DE) Molecular Similarity, vol. 2-3, Springer, 1998, 800 pages), TOC, pp. 243-244 provided.* *) NotOt1Ce: Subjubject to anyy d1Sclaimer,disclai theh term off thisthi Tatsuta et al. (Bioorg. Med. Chem. Lett. 15 (2005) 2265-2269).* patent is extended or adjusted under 35 Wermuth, The Practice of Medicinal Chemsitry, 2d ed. (2003), 768 U.S.C. 154(b) by 852 days. pages, chs. 9-10 provided.* CAPLUS Abstract of WO O3051837.* (21) Appl. No.: 12/109,479 * cited by examiner (22) Filed: Apr. 25, 2008 (65) Prior Publication Data Primary Examiner — Robert Havlin (74) Attorney, Agent, or Firm — Oblon, Spivak, US 2009/O 170783 A1 Jul. 2, 2009 McClelland, Maier & Neustadt, L.L.P. Related U.S. Application Data (60) Provisional application No. 60/914,424, filed on Apr. (57) ABSTRACT 27, 2007. The present invention provides novel tetrahydrocarbazole compounds according to formula (I) as ligands of G-protein (30) Foreign Application Priority Data coupled receptors (GPCR) which are useful in the treatment and/or prophylaxis of physiological and/or pathological con Apr. -

Yerevan State Medical University After M. Heratsi
YEREVAN STATE MEDICAL UNIVERSITY AFTER M. HERATSI DEPARTMENT OF PHARMACY Balasanyan M.G. Zhamharyan A.G. Afrikyan Sh. G. Khachaturyan M.S. Manjikyan A.P. MEDICINAL CHEMISTRY HANDOUT for the 3-rd-year pharmacy students (part 2) YEREVAN 2017 Analgesic Agents Agents that decrease pain are referred to as analgesics or as analgesics. Pain relieving agents are also called antinociceptives. An analgesic may be defined as a drug bringing about insensibility to pain without loss of consciousness. Pain has been classified into the following types: physiological, inflammatory, and neuropathic. Clearly, these all require different approaches to pain management. The three major classes of drugs used to manage pain are opioids, nonsteroidal anti-inflammatory agents, and non opioids with the central analgetic activity. Narcotic analgetics The prototype of opioids is Morphine. Morphine is obtained from opium, which is the partly dried latex from incised unripe capsules of Papaver somniferum. The opium contains a complex mixture of over 20 alkaloids. Two basic types of structures are recognized among the opium alkaloids, the phenanthrene (morphine) type and the benzylisoquinoline (papaverine) type (see structures), of which morphine, codeine, noscapine (narcotine), and papaverine are therapeutically the most important. The principle alkaloid in the mixture, and the one responsible for analgesic activity, is morphine. Morphine is an extremely complex molecule. In view of establish the structure a complicated molecule was to degrade the: compound into simpler molecules that were already known and could be identified. For example, the degradation of morphine with strong base produced methylamine, which established that there was an N-CH3 fragment in the molecule. -

Supplementary Digital Content REVIEW of ANXIETY MODELS USED in 2010-2011. Type of the Study. Here We Cathegorize the Study Accor
Supplementary digital content REVIEW OF ANXIETY MODELS USED IN 2010-2011. LEGEND Type of the study. Here we cathegorize the study according to its major aim or major finding as follows: anxiogenic effects, the compound had effects opposite to expectations; detection of anxiolytic effect, anxiolytic effects were noticed but no suggestion was formulated for use as anxiolytics; established anxiolytic, well-known anxiolytics studied for various reasons; no effect on anxiety, the compound did not fulfil expectations; mechanism, studies employing anxiolytic treatments but addressing developmental issues, neural mechanisms, drug interactions etc.; putative novel anxiolytic, the authors suggested the compound for anxiolytic drug development; (H), the tested compound was an herbal extract; (Ho), purified or synthesized compound of herbal origin. Anxiety tests. The tests listed in Table 1 were considered 'classical' for the reasons specified in the Abstract. Modifier. Tests are sometimes performed under unconventional conditions to induce heightened levels of anxiety and by this to icrease the translational value of the test. Procedures that altered (usually increased) anxiety levels normally shown in the given test were categorized as follows. chemical, hormonal or pharmacological treatments (e.g. ovarectomy, drug administration in adolescence, etc.); condition, modified testing conditions (e.g. high lighting, no habituation to the testing room, etc.); neural, treatments that affect neural functions (e.g. brain lesions, inhibited neurogenesis); selection, subjects from selection lines, specific strains, etc; stress, subjects exposed to stressors with long-lasting consequences (e.g. chronic immobility, drug withdrawal, etc); subject, the use of specific subjects classes (aged subjects, females, etc.); transgenic, the effects of drugs were studied in genetically engineered subjects. -

Download Product Insert (PDF)
PRODUCT INFORMATION Semax Item No. 27719 CAS Registry No.: 80714-61-0 N Formal Name: L-methionyl-L-α-glutamyl- N H S L-histidyl-L-phenylalanyl-L- H2N prolylglycyl-L-proline O Synonym: ACTH4-7-PGP N H N N O MF: C37H51N9O10S O FW: 813.9 H N OH N O H Purity: ≥95% N O O O Supplied as: A solid H Storage: -20°C O HO Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Semax is supplied as a solid. A stock solution may be made by dissolving the semax in the solvent of choice, which should be purged with an inert gas. Semax is soluble in organic solvents such as DMSO and dimethyl formamide. The solubility of semax in these solvents is approximately 5 and 1 mg/ml, respectively. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of semax can be prepared by directly dissolving the solid in aqueous buffers. The solubility of semax in PBS, pH 7.2, is approximately 10 mg/ml. We do not recommend storing the aqueous solution for more than one day. Description Semax is a synthetic peptide analog of adrenocorticotropic hormone (ACTH) (4-10) (Item No. 27106) that has neuroprotective, analgesic, and anxiolytic properties.1-3 In vivo, semax (0.3 mg/kg) reduces cortical nitric oxide (NO) production and the number of neurological disturbances, such as seizures, falling, and twisting, in a rat model of global ischemia.1 It decreases acetic acid-induced writhing and nociception in a hind paw compression test in mice when administered at doses ranging from 0.015 to 0.5 mg/kg.2 Semax also increases time spent in the open arms in the elevated plus maze, indicating anxiolytic activity, in a rat model of maternal deprivation-induced anxiety.3 References 1. -

Effects of Semax on Dopaminergic and Serotoninergic Systems of the Brain K
Doklady Biological Sciences, Vol. 394, 2004, pp. 1–3. Translated from Doklady Akademii Nauk, Vol. 394, No. 1, 2004, pp. 130–132. Original Russian Text Copyright © 2004 by Eremin, Kudrin, Grivennikov, Miasoedov, Rayevsky. PHYSIOLOGY Effects of Semax on Dopaminergic and Serotoninergic Systems of the Brain K. O. Eremin*, V. S. Kudrin*, I. A. Grivennikov**, Academician N. F. Miasoedov**, and K. S. Rayevsky* Received June 11, 2003 Short-term effects of the synthetic nootropic peptide in rats and simultaneously raises the intracellular level Semax on the content and metabolism of monoamines of dopamine in the striatum [6]. in the brain of C57/Bl6 mice and Sprague–Dawley rats The goal of this study was to examine the effects of were studied. Intraperitoneal injection of Semax at a Semax on the neurochemical parameters of the dopam- dose of 0.15 mg/kg caused an increase in the tissue con- inergic and serotoninergic systems of the animal brain. centrations of 5-hydroxyindoleacetic acid (5-HIAA) in Semax was administered at a dose of 0.15 mg per kilo- the hypothalamus and striatum of mice 0.5 and 2 h after gram body weight to ë57/Bl6 mice, and their hypothal- the injection. Using brain microdialysis, we detected amus and striatum were analyzed for tissue levels of increased levels of extracellular 5-HIAA in the striatum dopamine (DA) and its metabolites 3,4-dihydroxyphe- of freely moving rats 1 h after administration of 0.15 or nylacetic acid (DOPAC) and homovanillic acid (HVA), 0.6 mg/kg Semax. The effect lasted for an additional as well as serotonin (or 5-hydroxytriptamine, 5-HT) 3 h. -

(12) United States Patent (10) Patent No.: US 9,345,661 B2 Adler Et Al
USOO9345661 B2 (12) United States Patent (10) Patent No.: US 9,345,661 B2 Adler et al. (45) Date of Patent: May 24, 2016 (54) SUBCUTANEOUSANTI-HER2 ANTIBODY FOREIGN PATENT DOCUMENTS FORMULATIONS AND USES THEREOF CL 2756-2005 10/2005 CL 561-2011 3, 2011 (75) Inventors: Michael Adler, Riehen (CH); Ulla CL 269-2012 1, 2012 Grauschopf, Riehen (CH): CN 101163717 4/2008 Hanns-Christian Mahler, Basel (CH): CN 101370525 2, 2009 Oliver Boris Stauch, Freiburg (DE) EP O590058 B1 11, 2003 EP 1516628 B1 3, 2005 (73) Assignee: Genentech, Inc., South San Francisco, EP 1603541 11, 2009 JP 2007-533631 11, 2007 CA (US) JP 2008-5075.20 3, 2008 JP 2008-528638 T 2008 (*) Notice: Subject to any disclaimer, the term of this JP 2009-504142 2, 2009 patent is extended or adjusted under 35 JP 2009/055343 4/2009 U.S.C. 154(b) by 0 days. KR 2007-0068385 6, 2007 WO 93.21319 A1 10, 1993 WO 94/OO136 A1 1, 1994 (21) Appl. No.: 12/804,703 WO 97.048O1 2, 1997 WO 98.22136 5, 1998 (22) Filed: Jul. 27, 2010 WO 99.57134 11, 1999 WO 01.00245 A2 1, 2001 (65) Prior Publication Data WO 2004/078140 9, 2004 WO 2005/023328 3, 2005 US 2011 FOO44977 A1 Feb. 24, 2011 WO 2005/037992 A2 4/2005 WO 2005,117986 12/2005 (30) Foreign Application Priority Data WO WO 2005,117986 A2 * 12/2005 WO 2006/044908 A2 4/2006 Jul. 31, 2009 (EP) ..................................... 0.9167025 WO 2006/09 1871 8, 2006 WO 2007 O24715 3, 2007 (51) Int. -

Semax) Peptide at the Transcriptome Level Following Cerebral Ischaemia–Reperfusion in Rats
G C A T T A C G G C A T genes Article Novel Insights into the Protective Properties of ACTH(4-7)PGP (Semax) Peptide at the Transcriptome Level Following Cerebral Ischaemia–Reperfusion in Rats Ivan B. Filippenkov 1,* , Vasily V. Stavchansky 1, Alina E. Denisova 2, Vadim V. Yuzhakov 3, Larisa E. Sevan’kaeva 3, Olga Y. Sudarkina 1 , Veronika G. Dmitrieva 1, Leonid V. Gubsky 2, Nikolai F. Myasoedov 1, Svetlana A. Limborska 1 and Lyudmila V. Dergunova 1 1 Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia; [email protected] (V.V.S.); [email protected] (O.Y.S.); [email protected] (V.G.D.); [email protected] (N.F.M.); [email protected] (S.A.L.); [email protected] (L.V.D.) 2 Pirogov Russian National Research Medical University, Federal Center of Cerebrovascular Pathology and Stroke, Ministry of Health Care of Russian Federation, 117342 Moscow, Russia; [email protected] (A.E.D.); [email protected] (L.V.G.) 3 A. Tsyb Medical Radiological Research Center–Branch of the National Medical Research Radiological Center of the Ministry of Health of the Russian Federation, 249031 Obninsk, Russia; [email protected] (V.V.Y.); [email protected] (L.E.S.) * Correspondence: fi[email protected]; Tel.: +7-499-196-1858 Received: 16 May 2020; Accepted: 18 June 2020; Published: 22 June 2020 Abstract: Cerebral ischaemia is the most common cause of impaired brain function. Biologically active peptides represent potential drugs for reducing the damage that occurs after ischaemia. The synthetic melanocortin derivative, ACTH(4-7)PGP (Semax), has been used successfully in the treatment of patients with severe impairment of cerebral blood circulation. -

Wells Pharmacy Network Master Formulary 7-27-2020 Without Pricing
BHRT Pellets & Numbing Cream Medication Form Strength Pellet Size (mm) 6 mg (3 mm) 10 mg (3 mm) 12.5 mg (3 mm) Estradiol Pellet 15 mg (3 mm) 18 mg (3 mm) 20 mg (3 mm) 25 mg (3 mm) 50 mg (3 mm) Progesterone Pellet 100 mg (3 mm) 12.5 mg (3 mm) 25 mg (3 mm) 37.5 mg (3 mm) 50 mg (3 mm) Testosterone Pellet 62.5 mg (3 mm) 87.5 mg (3 mm) 100 mg (3 mm) 200 mg (4.5 mm) 100 mg / 4 mg (3 mm) Testosterone / Anastrozole Pellet 200 mg / 8 mg (4.5 mm) 200 mg / 20 mg (4.5 mm) Disposable - Each Plastic Tip (3.2 mm) Disposable - Stainless Steel Tip Each Trocar Kit for Pellet Insertion Trocar (3.2 mm & 4.5 mm) Reusable - Titanium Or Each Stainless Steel (3.2 mm & 4.5 mm) 3.2 mm Trocars are meant to be used with 3 mm Pellets & 4.5 mm Trocars are meant to be used with 4.5 mm Pellets Medication Form Strength Package Size Cream Benzocaine / Lidocaine/ Tetracaine 20% / 6% / 4% 120 gm Topi-Pump® Items marked in Yellow: (new items added with-in 90 days) Price effective July 27th, 2020 | Pricing Subject to Change | Products may be subject to additional fees based on availability 503B IV Therapy Medication Form Strength Package Size Arginine IV 100 mg / ml 30 mL Glutathione IV 200 mg / ml 30 mL Lipoic Acid (DL-Thioctic Acid) IV 2.5% (25 mg / ml) 30 mL Methylcobalamin IV 1000 mcg / ml 10 mL Pyridoxine HCL IV 100 mg / ml 30 mL Taurine IV 50 mg/ ml 30 mL Items marked in Yellow: (new items added with-in 90 days) Price effective July 27th, 2020 | Pricing Subject to Change | Products may be subject to additional fees based on availability Peptides Package Medication Form -

Nootropics- Memory Boosters
Harikumar K. et al. / Journal of Pharmaceutical Biology, 6(1), 2016, 14-19. Journal of Pharmaceutical Biology www.jpbjournal.com e-ISSN - 2249-7560 Print ISSN - 2249-7579 NOOTROPICS- MEMORY BOOSTERS K.Hari Kumar*, Mitta Srija, D.K.Sandeep, Ramisetty Davarika, Gunda Sai Mounica Department of Pharmacology, Sri Venkateswara College of Pharmacy, R.V.S.Nagar, Chittoor-517127, Andhra Pradesh, India. ABSTRACT Nootropics also called as smart drug, memory enhancers, neuron enhancers, cognitive enhancers and intelligent enhancers are drugs, supplements, neutraceuticals and functional foods that provide one or more aspects of mental function. Specific effect can include improvement to working memory motivation or attention. Nootropics drugs are able to promote, enhance and protect cognitive functions. As cognition is the typically human higher activity of brain, nootropic concept looked quite appealing for scores of people dreaming to enjoy better and longer lasting mental activity and for drug maker keen to produce such enviable products. There are large number of drugs which can be used as nootropic agents and help to enhance memory of people. Nootropics offer lot of benefits for cognitive aptitude and brain health. Nootropics used for the treatment of Alzheimer’s disease, Parkinson’s disease and Huntington’s disease, dementia and cognitive symptoms of schizophrenia. Keywords: Nootropics, Huntington’s disease, Dementia, Alzheimer’s disease. INTRODUCTION It is derived from Greek words “NOOS –Mind” But non-ADHD medications and supplements are more “Tropein – Turn/Bend”. They are also called as memory frequently used for performance enhancement. Many enhancers, Smart nutrients, Cerebroactive drugs and individuals use these drugs for performance enhancement cognition enhancers. -

Impact on Patient Satisfaction with Methadone
INFLUENCING FACTORS ON METHADONE PHARMACOLOGY: IMPACT ON PATIENT SATISFACTION WITH METHADONE MAINTENANCE TREATMENT by Alexander Khaled Elkader A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Graduate Department of Pharmaceutical Sciences University of Toronto © Copyright by Alexander Khaled Elkader (2009) i Influencing Factors on Methadone Pharmacology: Impact on Patient Satisfaction with Methadone Maintenance Treatment. Alexander Khaled Elkader Doctor of Philosophy Department of Pharmaceutical Sciences University of Toronto 2009 Abstract: The methadone maintenance treatment population suffers from high rates of comorbid psychiatric and substance use disorders. Despite a more than 40-year treatment history, not all patients are satisfied with methadone treatment and more than half of the patients complain of significant inter-dose withdrawal at least some of the time. The objectives of this research were to investigate the pharmacological response to methadone under the influence of comorbid major depressive disorder and smoking; and to identify factors other than physical withdrawal symptoms that can differentiate patients based on their complaints of dissatisfaction with treatment. In Study 1, seven depressed methadone maintenance patients experienced more opioid withdrawal symptomatology over a 24- hour methadone-dosing interval than 10 nondepressed methadone patients. Depression severity was significantly correlated with trough opioid withdrawal severity. This suggests that depression or depressive symptoms are related to reported opioid withdrawal. In Study 2, many factors other than physical opioid withdrawal symptoms were able to differentiate patients who were satisfied with treatment (holders, n=25), partially satisfied with treatment (partial holders, n=35), and not satisfied with treatment (nonholders, n=30). Results suggested that these patient satisfaction groups cluster differently depending on physical opioid withdrawal, mood, psychological distress, and ii personality. -

Selank and Semax As Potential Hepatoprotectors in Medical
Research Results in Pharmacology 5(4): 33–39 UDC: 615.331 DOI 10.3897/rrpharmacology.5.38769 Research Article Selank and semax as potential hepatoprotectors in medical treatment of tuberculosis Alexey K. Petrovsky1, Nikolay A. Smirnov1, Vladimir P. Vdovichenko2, Tatiana B. Fedorova1, Edgar E. Kerbenev1, Vladimir N. Fedorov1 1 Yaroslavl State Medical University, 5 Revolyutsionnaya St., Yaroslavl 150000, Russian Federation 2 Grodno State Medical University, 80 Gorky St., Grodno 230009, Republic of Belarus Corresponding author: Alexey K. Petrovsky ([email protected]) Academic editor: Mikhail Korokin ♦ Received 2 August 2019 ♦ Accepted 24 October 2019 ♦ Published 16 December 2019 Citation: Petrovsky AK, Smirnov NA, Vdovichenko VP, Fedorova TB, Kerbenev EE, Fedorov VN (2019) Selank and semax as potential hepatoprotectors in medical treatment of tuberculosis. Research Results in Pharmacology 5(4): 33–39. https://doi. org/10.3897/rrpharmacology.5.38769 Abstract Introduction: Drug-induced hepatitis is common in clinical practice. This problem is particularly relevant in the treat- ment of tuberculous infection, because for this purpose, up to 5–6 hepatotoxic drugs are used simultaneously for a long time, which often (in 15–20% of cases) leads to medical liver lesion. To protect the liver, Semax and Selank are offered – drugs of regulatory peptides group. Materials and Methods: The research was conducted on 96 outbred white male rats weighing 180–220 g. The experi- mental group included about 10 animals. Drug-induced hepatitis was simulated through the combined 21-day adminis- tration of isoniazid, rifampicin and ethanol. Semax and Selank, as well as Essentiale N and Mexidol (comparison drugs) were administered once a day during the experiment. -
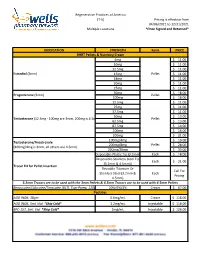
Formularies Pricing Sheet
Regenerative Practices of America (T-5) Pricing is effective from 04/06/2021 to 12/31/2021 Multiple Locations *Once Signed and Returned* MEDICATION STRENGTH Form PRICE BHRT Pellets & Numbing Cream 6mg $ 11.00 10mg $ 11.00 12.5mg $ 11.00 Estradiol (3mm) 15mg Pellet $ 11.00 18mg $ 11.00 20mg $ 11.00 25mg $ 11.00 50mg $ 18.00 Progesterone (3mm) Pellet 100mg $ 18.00 12.5mg $ 11.00 25mg $ 11.00 37.5mg $ 11.00 50mg $ 13.00 Testosterone (12.5mg - 100mg are 3mm, 200mg is 4.5mm) Pellet 62.5mg $ 13.00 87.5mg $ 14.00 100mg $ 16.00 200mg $ 21.00 100mg/4mg $ 19.00 Testosterone/Anastrozole 200mg/8mg Pellet $ 28.00 (100mg/4mg is 3mm, all others are 4.5mm) 200mg/20mg $ 33.00 Disposable-Plastic Tip (3.2mm) Each $ 18.00 Disposable-Stainless Steel Tip Each $ 21.00 (3.2mm & 4.5mm)) Trocar Kit for Pellet Insertion Reusable-Titanium Or Call For Stainless Steel (3.2mm & Each Pricing 4.5mm) 3.2mm Trocars are to be used with the 3mm Pellets & 4.5mm Trocars are to be used with 4.5mm Pellets Benzocaine/Lidocaine/Teracaine (BLT), Topi-Pump, 120gm 20%/6%/4% Cream $ 67.00 Peptides AOD 9604, 30gm 0.6mg/mL Cream $ 118.00 AOD 9604, 5mL Vial *Ship Cold* 1.2mg/mL Injectable $ 118.00 BPC-157, 5mL Vial *Ship Cold* 2mg/mL Injectable $ 126.00 Regenerative Practices of America (T-5) Pricing is effective from 04/06/2021 to 12/31/2021 Multiple Locations *Once Signed and Returned* 250mcg (0.25 mg) Capsule $ 74.00 BPC-157, Qty #30 *Ship Cold* 500mcg (0.5mg) Capsule $ 105.00 1mg Capsule $ 120.00 1mg/1mg/mL Injectable $ 55.00 CJC-1295/Ipamorelin, 2mL Vial *Ship Cold* 2mg/1mg/mL Injectable