Improve Your Galaxy Text Life: the Query Tabular Tool[Version 1; Peer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Release 0.0.1 Alan Mitchell
bmon Documentation Release 0.0.1 Alan Mitchell Sep 16, 2021 Users 1 User Introduction 3 2 System Administrator Introduction5 3 Developer Introduction 7 4 Contact Information 9 4.1 User Introduction.............................................9 4.2 System Administrator Introduction................................... 10 4.3 How to Install BMON on a Web Server................................. 13 4.4 How to Install BMON on a Local Web Server.............................. 18 4.5 Add Buildings and Sensors....................................... 27 4.6 Sharing BMON across Multiple Organizations............................. 40 4.7 Setting Up Sensors to Post to BMON.................................. 41 4.8 Multi-Building Charts.......................................... 60 4.9 Sensor Alerts............................................... 68 4.10 Creating a Dashboard.......................................... 74 4.11 Transform Expressions.......................................... 76 4.12 Calculated Fields............................................. 80 4.13 Periodic Scripts.............................................. 92 4.14 How to Create Custom Jupyter Notebook Reports........................... 107 4.15 Custom Reports............................................. 108 4.16 Backing Up and Analyzing Data from the System........................... 111 4.17 System Performance with High Loading................................ 113 4.18 Using CSV Transfer........................................... 115 4.19 Developer Introduction......................................... -

Web Vulnerabilities (Level 1 Scan)
Web Vulnerabilities (Level 1 Scan) Vulnerability Name CVE CWE Severity .htaccess file readable CWE-16 ASP code injection CWE-95 High ASP.NET MVC version disclosure CWE-200 Low ASP.NET application trace enabled CWE-16 Medium ASP.NET debugging enabled CWE-16 Low ASP.NET diagnostic page CWE-200 Medium ASP.NET error message CWE-200 Medium ASP.NET padding oracle vulnerability CVE-2010-3332 CWE-310 High ASP.NET path disclosure CWE-200 Low ASP.NET version disclosure CWE-200 Low AWStats script CWE-538 Medium Access database found CWE-538 Medium Adobe ColdFusion 9 administrative login bypass CVE-2013-0625 CVE-2013-0629CVE-2013-0631 CVE-2013-0 CWE-287 High 632 Adobe ColdFusion directory traversal CVE-2013-3336 CWE-22 High Adobe Coldfusion 8 multiple linked XSS CVE-2009-1872 CWE-79 High vulnerabilies Adobe Flex 3 DOM-based XSS vulnerability CVE-2008-2640 CWE-79 High AjaxControlToolkit directory traversal CVE-2015-4670 CWE-434 High Akeeba backup access control bypass CWE-287 High AmCharts SWF XSS vulnerability CVE-2012-1303 CWE-79 High Amazon S3 public bucket CWE-264 Medium AngularJS client-side template injection CWE-79 High Apache 2.0.39 Win32 directory traversal CVE-2002-0661 CWE-22 High Apache 2.0.43 Win32 file reading vulnerability CVE-2003-0017 CWE-20 High Apache 2.2.14 mod_isapi Dangling Pointer CVE-2010-0425 CWE-20 High Apache 2.x version equal to 2.0.51 CVE-2004-0811 CWE-264 Medium Apache 2.x version older than 2.0.43 CVE-2002-0840 CVE-2002-1156 CWE-538 Medium Apache 2.x version older than 2.0.45 CVE-2003-0132 CWE-400 Medium Apache 2.x version -

Before We Start…
Before we start… This is the Introduction to Databases Design and Implementation workshop • Download material: dartgo.org/db-design • Poll / Interactive questions: dartgo.org/poll • Optional software: https://dev.mysql.com/downloads/workbench/ • More info: rc.dartmouth.edu Introduction to Database Design and Implementation Christian Darabos, Ph.D. [email protected] Slides download: dartgo.org/db-design Overview • introduction to Databases and this workshop • development/production environments • tools (admin, browse, query, etc.) • DB design, UML and case study (http://www.datanamic.com/support/lt-dez005-introduction-db-model ing.html) • port model into MySQL Workbench Right-click > Open link in new window To keep open slides and poll dartgo.org/poll Research Computing Introduction • Research Computing service offering • Definition of a Relational Database • Overview of this workshop Right-click > Open link in new window To keep open slides and poll dartgo.org/poll Definition of a Relational Database (SQL) • a database type structured to recognize relations among stored items of information • designed to store text, dates/times, integers, floating-point number • implemented as a series of tables Mental Model • Think of a database as a set of spreadsheets • Each spreadsheet (or table) represents a type of entity (person, object, concept, etc.) • Better than Excel because it also models the relationship between the entities Why use a Relational Database • concurrent (simultaneous) read and write • powerful selecting, filtering and sorting cross-referencing tables • large quantity of structured storage and standardized distribution • minimize post-processing (simple analytics tools pre-implemented) • automate using any scripting and programming languages (R, Matlab, Python, C++, Java, PHP) • web-proof SQL vs. -

Iot Energy Monitoring of a Refrigeration Installation
WEENTECH Proceedings in Energy 6 (2020) 88-98 Page | 88 5th Virtual International Conference on Energy, Environment and Economics, ICEEE2020, 18-19 August 2020 IoT energy monitoring of a refrigeration installation José Uslenghi, Angel Sapena-Bano*, Manuel Pineda-Sanchez, Jordi Burriel-Valencia, Ruben Puche-Panadero, Javier Martinez-Roman Institute for Energy Engineering, Universitat Politècnica de València, 46022 València, Spain *Corresponding author’s mail: [email protected] Abstract This paper presents an application for the energy monitoring of commercial refrigeration facility, based on the concept and technology of the Internet of Things. The purpose of this application is to offer support to the activities of energy audits ant to the implementation of energy management systems, where the energy consumption of a facility must be characterized, or the saving measures applied must be supervised. Among the main features offered by this application are the possibility of remotely monitoring the operating conditions of a refrigeration cycle, as well as its energy consumption. Likewise, its Internet connectivity allows the monitoring system to be scaled up and to group the measurements of several refrigeration installations, which transforms this application into a powerful tool for benchmarking. Internet access also allows notifications via e-mail under particular conditions of the operation of any of the facilities. On the other hand, the application has the capacity to collect and to store in a database the measurements taken by the field devices, allowing the generation of historical reports showing the evolution of the operation of the installation. The main component of this application is the industrial gateway, SIMATIC IOT2040, which is used to read industrial communication buses and to connect to the Internet. -

Pacsystems* IC695CPL410 Rx3i 1.2Ghz 64MB Rackless CPU W/Linux Quick Start Guide GFK-3053 July 2018
GE Automation & Controls Programmable Control Products PACSystems* IC695CPL410 RX3i 1.2GHz 64MB Rackless CPU w/Linux Quick Start Guide GFK-3053 July 2018 For Public Disclosure Contents 1. User Features........................................................................................................... 1 1.1. Switches ........................................................................................................... 5 1.2. Displays and Indicators (LEDs) ......................................................................... 5 1.3. USB Ports ......................................................................................................... 8 1.4. Front-Panel Ethernet Ports ............................................................................... 8 1.5. Serial COM Port ..............................................................................................10 1.6. Video Display Port ...........................................................................................10 1.7. Linux Port ........................................................................................................10 1.8. Energy Pack Connector ...................................................................................11 1.9. Input Power Connector ....................................................................................11 1.10. Removable Data Storage Device (RDSD) ........................................................11 2. Hardware Installation ......................................................................................... -

Demystifying Africa's Cyber Security Poverty Line
Demystifying Africa’s Cyber Security Poverty Line Tanzania Chapter The Africa Cyber Immersion Centre is a state-of-the-art research, innovation and training facility that seeks to address Africa’s ongoing and long-term future needs through unique education, training, research, and practical applications. For more information Serianu Limited contact: [email protected] http://www.serianu.com Tanzania Cyber Security 3 Report Content Editor’s Note and Acknowledgement Cost of Cyber Crime We are excited to finally publish the 2nd edition of Tanzania We estimate that cyber-attacks cost Tanzanian 4 Cyber Security Report 2017. 54 businesses around $99.5 million a year. Foreword Sector Ranking in 2017 2017 has seen a jump in cyberattacks or cybercrimes. Cyber security is no longer a concern fof the Ransomware, DDoS attacks, data breaches were all financial & banking sectors only. 7 synonymous with 2017. 56 Executive Summary Home Security It is in our own best interests to make sure everyone – from the young to the old, on The global landscape of cyber threats is quickly changing. snapchat, facebook and twitter - know and 9 60 practice basic security habits. Top Trends Africa Cyber Security Framework We analysed incidents that occurred in 2017 and compiled Attackers are now launching increasingly a list of top trends that had a huge impact on the economic sophisticated attacks on everything from and social well-being of organisations and Tanzanian citizens. business critical infrastructure to everyday 13 69 devices such as mobile phones. Top Priorities for 2018 Appendixes 19 We have highlighted key priorities for 2018. 71 Cyber Intelligence Statistics, Analysis, & Trends References We have monitored organisations’ network for malware and cyber threat attacks such as brute-force attacks 25 against the organisation’s servers. -

Raspberry Pi Server Essentials
Raspberry Pi Server Essentials Transform your Raspberry Pi into a server for hosting websites, games, or even your Bitcoin network Piotr J. Kula BIRMINGHAM - MUMBAI Raspberry Pi Server Essentials Copyright © 2014 Packt Publishing All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, without the prior written permission of the publisher, except in the case of brief quotations embedded in critical articles or reviews. Every effort has been made in the preparation of this book to ensure the accuracy of the information presented. However, the information contained in this book is sold without warranty, either express or implied. Neither the author, nor Packt Publishing, and its dealers and distributors will be held liable for any damages caused or alleged to be caused directly or indirectly by this book. Packt Publishing has endeavored to provide trademark information about all of the companies and products mentioned in this book by the appropriate use of capitals. However, Packt Publishing cannot guarantee the accuracy of this information. First published: February 2014 Production Reference: 1030214 Published by Packt Publishing Ltd. Livery Place 35 Livery Street Birmingham B3 2PB, UK. ISBN 978-1-78328-469-6 www.packtpub.com Cover Image by Piotr J. Kula ([email protected]) Credits Author Project Coordinator Piotr J. Kula Sageer Parkar Reviewers Proofreaders Teemu Lätti Maria Gould Warren Myers Paul Hindle Acquisition Editor Indexers Kunal Parikh Monica Ajmera Mehta Tejal Soni Commissioning Editor Manasi Pandire Graphics Ronak Dhruv Technical Editors Shubhangi Dhamgaye Production Coordinator Nachiket Vartak Shantanu Zagade Copy Editors Cover Work Alisha Aranha Shantanu Zagade Roshni Banerjee Gladson Monteiro Adithi Shetty About the Author Piotr J. -
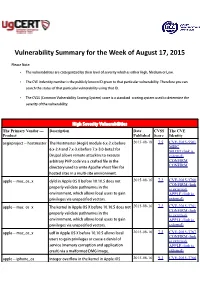
Vulnerability Summary for the Week of August 17, 2015
Vulnerability Summary for the Week of August 17, 2015 Please Note: • The vulnerabilities are cattegorized by their level of severity which is either High, Medium or Low. • The !" indentity number is the #ublicly $nown %& given to that #articular vulnerability. Therefore you can search the status of that #articular vulnerability using that %&. • The !'S (Common !ulnerability 'coring System) score is a standard scoring system used to determine the severity of the vulnerability. High Severity Vulnerabilities The Primary Vendor --- Description Date CVSS The CVE Product Published Score Identity aegir#roject ++ hostmaster The Hostmaster (,egir) module -.x+/.x before 2015-08-18 7.5 CVE-2015-5501 MISC -.x+/.4 and 1.x+2.x before 1.x+2.3+beta/ for MLIST (link is Dru#al allows remote attac$ers to e.ecute external) arbitrary PHP code via a crafted file in the CONFIRM CONFIRM directory used to write ,#ache vhost files for hosted sites in a multi+site environment. a##le ++ mac4os4. dyld in ,##le 5' 6 before 73.13.5 does not 2015-08-16 7.2 CVE-2015-3760 CONFIRM (link #ro#erly validate #athnames in the is external) environment, which allows local users to gain APPLE (link is #rivileges via uns#ecified vectors. external) a##le ++ mac4os4. The $ernel in ,##le 5' 6 before 73.13.8 does not 2015-08-16 7.2 CVE-2015-3761 CONFIRM (link #ro#erly validate #athnames in the is external) environment, which allows local users to gain APPLE (link is #rivileges via uns#ecified vectors. external) a##le ++ mac4os4. udf in ,##le 5' 6 before 73.73.5 allows local 2015-08-16 7.2 CVE-2015-3767 CONFIRM (link users to gain #rivileges or cause a denial of is external) service (memory corru#tion and a##lication APPLE (link is crash) via a malformed DM9 image. -
“Embedded” Con Sqlite
Bandiera Roberto – Progettazione e Sviluppo Web con CodeIgniter 3 – marzo 2019 Appendice 5 – Uso di un database “embedded” con SQLite La libreria SQLite (https://www.sqlite.org ) consente di creare e gestire un database di tipo relazionale incorporato nella propria applicazione. Il database è costituito da un singolo file che viene agevolmente inserito all’interno delle cartelle dell’applicazione e trasportato assieme alla stessa. Per poter utilizzare il database non c’è bisogno di alcuna installazione o configurazione, ne’ di avere a disposizione un apposito servizio software, aggiuntivo rispetto all’applicazione stessa. Con SQLite si crea, tipicamente, un database pensato per un uso personale, tuttavia SQLite è anche in grado di gestire l’accesso concorrente di più utenti, applicare i vincoli di integrità del database, garantire transazioni con proprietà ACID (Atomicità, Consistenza, Isolamento, Persistenza). Le prestazioni, in termini di numero di accessi simultanei al database, sono inferiori a quelle di un database gestito da un vero server, come MySQL, SQLServer, Oracle; tuttavia SQLite può venire impiegato, oltre che per una archiviazione di dati per uso personale, come quella inserita nei browser web, anche per realizzare un prototipo funzionante di una applicazione multiutente o per effettive applicazioni web di medio-piccole dimensioni, dove non si ha a disposizione un servizio di DBMS o non se ne vogliono sostenere i costi! Si ha anche il vantaggio di poter spostare agevolmente l’applicazione con il suo database da un server web ad un altro. Per una discussione sulla convenienza di uso di SQLite rispetto ad una soluzione client-server tradizionale si veda ad esempio “Usi appropriati di SQLite” (https://code-examples.net/it/docs/sqlite/whentouse ). -

CS50 Quiz Review
CS50 Quiz Review November 13, 2017 Info http://docs.cs50.net/2017/fall/quiz/about.html • 48-hour window in which to take the quiz. • You should require much less than that; expect an appropriately-scaled down version of the Test. • Released Tue 11/14 at noon, due via submit50 Thu 11/16 at noon. • Be sure to run update50 in your IDE before submitting! • Submitting seven minutes late is equivalent to not submitting at all; don’t wait until the last possible second. Info http://docs.cs50.net/2017/fall/quiz/about.html • Any topic that we have covered in the entire course is fair game. • The quiz will nonetheless be focused primarily on content from Weeks 6 through 10, inclusive, and Problem Sets 6 through 8, inclusive. Resources • Consult the syllabus for a guide of topics. • Review lecture notes. • Review lecture source code. • Review lecture slides. • (Re)watch lecture videos. • Review problem set specifications, distribution code, and sample solutions. Resources • Office hours • Tonight (here!) from 8pm – 10pm. Resources • Office hours • Tonight (here!) from 8pm – 10pm. • No office hours during the Test (11/14 through 11/16) or indeed for the remainder of the semester! Resources • CS50 Discourse • You may post questions through the end of the day today. • You may not post questions on Discourse from Tue 11/14 through Thu 11/16. • Staff will not respond to any questions during this time, but will monitor the forum. Resources • CS50 Discourse • You may post questions through the end of the day today. • You may not post questions on Discourse from Tue 11/14 through Thu 11/16. -

Black Falcon Phpliteadmin 취약점 분석
Confidential Black Falcon phpLiteAdmin 취약점 분석 2016. 05. by R2dF1r2 (All For One One For All ) 해당 문건은 대외비 문건으로, 팀 구성원만 열람 하도록 함. 해당 문건은 젂자 자산으로, 팀 내 관리하며 젂자 자료로 저장 관리함. 해당 문건은 팀장급 이상 열람 가능하며, 하드 카피는 불허 함. 해당 문건의 모든 권한은 BlackFalcon 대장에게 있음. 보안등급 Confidential BlackFalcon 문서번호 BF-2016-0005 phpLiteAdmin vulnerabiliteis 작성일자 2016-05-16 목차 1. phpLiteAdmin 소개 .............................................................................. 3 1.1 phpLiteAdmin 소개 ..................................................................................................................... 3 2. phpLiteAdmin 1.9.6 Multiple Vulnerabilities .................................. 4 2.1 취약점 소개 ................................................................................................................................... 4 2.2 CSRF 취약점 .................................................................................................................................. 4 3. phpLiteAdmin 1.9.6 CSRF 취약점 ...................................................... 4 3.1 Test 환경 ....................................................................................................................................... 4 3.2 phpLiteAdmin 1.9.6 CSRF 취약점 재현 .................................................................................... 6 2 보안등급 Confidential BlackFalcon 문서번호 BF-2016-0005 phpLiteAdmin vulnerabiliteis 작성일자 2016-05-16 1. phpLiteAdmin 소개 1.1 phpLiteAdmin 소개 phpLiteAdmin 은 웹사이트 형태로 SQLite를 관리할 수 있도록 도와주는 프로그램으로 php파일 하나로 구성된 싱글소스 프로그램이며 설치가 필요 없어 쉽게 사용할 수 있다. phpLiteAdmin -

Sweetie Framework: Simple but Practical Web Application Development Environment
1 Sweetie Framework: Simple but Practical Web Application Development Environment Motoki Miura Department of Basic Sciences, Faculty of Engineering, Kyushu Institute of Technology 1-1 Sensui, Tobata, Kitakyushu, Fukuoka, 804-8550 JAPAN Email: [email protected] Web: http://ist.mns.kyutech.ac.jp/ Abstract—In order to build a web application, understanding of the programming language, database, SQL, and HTTP is indispensable. Therefore, for beginners, the required skills of the web application development are higher than that of the web page authoring. We propose Sweetie framework that can easily learn basic concept of simple web applications and construct them. Sweetie framework is designed to work with Sweetie, a lightweight web development editor that runs on a web browser. By utilizing the Sweetie framework, novice developers can easily handle, retrieve, and display data in the database. In addition, the Sweetie framework provides user authentication and user registration function for practical applications. We have conducted a web application development lecture using Sweetie and Sweetie framework. Index Terms—Web-based Creative Task, Novice Web Developer, Web Framework, IDE ✦ 1INTRODUCTION Authoring web documents is one of the popular ac- than HTML, the learner can develop a web application in tivities in introductory computer literacy lecture courses. a short period of time. For improving usability, we bundled By authoring web documents, learners can understand the the Sweetie framework with web-based online editor named concept of markup languages, structure of hypertext, and Sweetie. Therefore, the teacher can provide optimized web representation of the web pages. Developing web appli- IDEs for novice learners. cations is an advanced course of the static web document authoring.