Complete Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Complementary DNA Microarray Analysis of Chemokines and Their Receptors in Allergic Rhinitis RX Zhang,1 SQ Yu,2 JZ Jiang,3 GJ Liu3
RX Zhang, et al ORIGINAL ARTICLE Complementary DNA Microarray Analysis of Chemokines and Their Receptors in Allergic Rhinitis RX Zhang,1 SQ Yu,2 JZ Jiang,3 GJ Liu3 1 Department of Otolaryngology, Huadong Hospital, Fudan University, Shanghai, China 2 Department of Otolaryngology , Jinan General Hospital of PLA, Shandong, China 3 Department of Otolaryngology, Changhai Hospital, Second Military Medical University, Shanghai, China ■ Abstract Objective: To analyze the roles of chemokines and their receptors in the pathogenesis of allergic rhinitis by observing the complementary DNA (cDNA) expression of the chemokines and their receptors in the nasal mucosa of patients with and without allergic rhinitis, using gene chips. Methods: The total RNAs were isolated from the nasal mucosa of 20 allergic rhinitis patients and purifi ed to messenger RNAs, and then reversely transcribed to cDNAs and incorporated with samples of fl uorescence-labeled with Cy5-dUPT (rhinitis patient samples) or Cy3- dUTP (control samples of nonallergic nasal mucosa). Thirty-nine cDNAs of chemokines and their receptors were latticed into expression profi le chips, which were hybridized with probes and then scanned with the computer to study gene expression according to the different fl uorescence intensities. Results: The cDNAs of the following chemokines were upregulated: CCL1, CCL2, CCL5, CCL7, CCL8, CCL11, CCL13, CCL14, CCL17, CCL18, CCL19, CCL24, and CX3CL1 in most of the allergic rhinitis sample chips. CCR2, CCR3, CCR4, CCR5, CCR8 and CX3CR1 were the highly expressed receptor genes. Low expression of CXCL4 was found in these tissues. Conclusion: The T helper cell (TH) immune system is not well regulated in allergic rhinitis. -

Overexpression of Microrna-155 Suppresses Chemokine Expression Induced by Interleukin-13 in BEAS-2B Human Bronchial Epithelial Cells
Allergology International 65 (2016) S17eS23 Contents lists available at ScienceDirect Allergology International journal homepage: http://www.elsevier.com/locate/alit Original article Overexpression of microRNA-155 suppresses chemokine expression induced by Interleukin-13 in BEAS-2B human bronchial epithelial cells * Satoshi Matsukura a, , Yuki Osakabe a, Ayaka Sekiguchi a, Daisuke Inoue a, Yusuke Kakiuchi a, Toshitaka Funaki a, Yohei Yamazaki a, Hiromi Takayasu a, Hidetsugu Tateno a, Eisuke Kato a, Aya Wakabayashi a, Makoto Hayashi a, Gen Ishii a, b, Fumihiro Yamaguchi a, Yutaka Tsuchiya a, Keita Kasahara b, Hironori Sagara c, Fumio Kokubu a a Department of Respiratory Internal Medicine, Showa University Fujigaoka Hospital, Kanagawa, Japan b Respiratory Disease Center, Showa University Northern Yokohama Hospital, Kanagawa, Japan c Department of Internal Medicine, Division of Allergy and Respiratory Medicine, Showa University School of Medicine, Tokyo, Japan article info abstract Article history: Background: MicroRNAs are non-coding small RNAs that regulate expression of target genes by binding Received 22 January 2016 to 30 untranslated regions. In this study, we used bronchial epithelial cells to investigate in vitro the role Received in revised form of the microRNA miR-155 in the expression of chemokines associated with airway inflammation. miR- 23 April 2016 155 has previously been reported to regulate allergic inflammation. Accepted 30 April 2016 Methods: BEAS-2B bronchial epithelial cells were cultured and transfected with mimic or inhibitor oli- Available online 3 August 2016 gonucleotides to overexpress or downregulate miR-155, as confirmed by real-time PCR. Cells were then stimulated with tumor necrosis factor-alpha, interleukin-13 (IL-13), and a double stranded RNA that Keywords: Asthma binds Toll-like receptor 3. -

Th22 Cells Represent a Distinct Human T Cell Subset Involved in Epidermal Immunity and Remodeling
Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling Stefanie Eyerich, … , Carsten B. Schmidt-Weber, Andrea Cavani J Clin Invest. 2009;119(12):3573-3585. https://doi.org/10.1172/JCI40202. Research Article Immunology Th subsets are defined according to their production of lineage-indicating cytokines and functions. In this study, we have identified a subset of human Th cells that infiltrates the epidermis in individuals with inflammatory skin disorders and is characterized by the secretion of IL-22 and TNF-α, but not IFN-γ, IL-4, or IL-17. In analogy to the Th17 subset, cells with this cytokine profile have been named the Th22 subset. Th22 clones derived from patients with psoriasis were stable in culture and exhibited a transcriptome profile clearly separate from those of Th1, Th2, and Th17 cells; it included genes encoding proteins involved in tissue remodeling, such as FGFs, and chemokines involved in angiogenesis and fibrosis. Primary human keratinocytes exposed to Th22 supernatants expressed a transcriptome response profile that included genes involved in innate immune pathways and the induction and modulation of adaptive immunity. These proinflammatory Th22 responses were synergistically dependent on IL-22 and TNF-α. Furthermore, Th22 supernatants enhanced wound healing in an in vitro injury model, which was exclusively dependent on IL-22. In conclusion, the human Th22 subset may represent a separate T cell subset with a distinct identity with respect to gene expression and function, present within the epidermal layer in inflammatory skin diseases. Future strategies directed against the Th22 subset may be of value in chronic inflammatory skin disorders. -

STAT6-Dependent Fashion CCL23 Expression Is Induced by IL-4 in A
CCL23 Expression Is Induced by IL-4 in a STAT6-Dependent Fashion Hermann Novak, Anke Müller, Nathalie Harrer, Claudia Günther, Jose M. Carballido and Maximilian Woisetschläger This information is current as of September 28, 2021. J Immunol 2007; 178:4335-4341; ; doi: 10.4049/jimmunol.178.7.4335 http://www.jimmunol.org/content/178/7/4335 Downloaded from References This article cites 47 articles, 20 of which you can access for free at: http://www.jimmunol.org/content/178/7/4335.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 28, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology CCL23 Expression Is Induced by IL-4 in a STAT6-Dependent Fashion Hermann Novak, Anke Mu¨ller, Nathalie Harrer, Claudia Gu¨nther, Jose M. Carballido, and Maximilian Woisetschla¨ger1 The chemokine CCL23 is primarily expressed in cells of the myeloid lineage but little information about its regulation is available. -

Association of Nerve Growth Factor, Chemokine (CC Motif)
Acta Derm Venereol 2013; 93: 144–149 INVESTIGATIVE REPORT Association of Nerve Growth Factor, Chemokine (C-C motif) Ligands and Immunoglobulin E with Pruritus in Cutaneous T-cell Lymphoma Hiraku SUGA, Makoto SUGAYA, Tomomitsu MIYAGAKI, Hanako Ohmatsu, Hideki Fujita, Shinji KAGAMI, Yoshihide ASANO, Yayoi TADA, Takafumi KADONO and Shinichi Sato Department of Dermatology, Faculty of Medicine, University of Tokyo, Tokyo, Japan Many patients with cutaneous T-cell lymphoma (CTCL) skin (6). The level of NGF is increased in inflamed experience severe pruritus. This study evaluated serum skin and pretreatment with anti-NGF serum prevents levels of nerve growth factor (NGF) and brain-derived increased neuropeptide expression at a neuronal level neurotrophic factor (BDNF) in patients with CTCL. (7). BDNF is produced by a number of immune cells, Although serum NGF and BDNF levels in patients with including mast cells, B cells, T-helper (Th) 2 cells and CTCL were not significantly higher than in healthy con- eosinophils (8–10). Studies have suggested that NGF trols, serum NGF levels in patients with Sézary syndrome and BDNF play important roles in the pathogenesis of were higher than in those with mycosis fungoides and in atopic dermatitis (AD). Serum levels of NGF and BDNF healthy controls. Enhanced NGF expression by kerati- are increased, providing a useful indicator of disease nocytes and increased dermal nerve fibres were detected activity in patients with AD (5, 11–13). in lesional skin of subjects with Sézary syndrome. Cor- This study evaluated serum NGF and BDNF levels relations between pruritus in CTCL and serum levels of in patients with CTCL in order to elucidate the factors NGF, BDNF, chemokine (C-C motif) ligand 1 (CCL1), related to pruritus in CTCL. -

An Introduction to Chemokines and Their Roles in Transfusion Medicine
Vox Sanguinis (2009) 96, 183–198 © 2008 The Author(s) REVIEW Journal compilation © 2008 Blackwell Publishing Ltd. DOI: 10.1111/j.1423-0410.2008.01127.x AnBlackwell Publishing Ltd introduction to chemokines and their roles in transfusion medicine R. D. Davenport Blood Bank and Transfusion Service, University of Michigan Health System, Ann Arbor, MI, USA Chemokines are a set of structurally related peptides that were first characterized as chemoattractants and have subsequently been shown to have many functions in homeostasis and pathophysiology. Diversity and redundancy of chemokine function is imparted by both selectivity and overlap in the specificity of chemokine receptors for their ligands. Chemokines have roles impacting transfusion medicine in haemat- opoiesis, haematologic malignancies, transfusion reactions, graft-versus-host disease, and viral infections. In haematopoietic cell transplantation, chemokines are active in mobilization and homing of progenitor cells, as well as mediating T-cell recruitment in graft-versus-host disease. Platelets are rich source of chemokines that recruit and activate leucocytes during thrombosis. Important transfusion-transmissible viruses such as cytomegalovirus and human immunodeficiency virus exploit chemokine receptors to evade host immunity. Chemokines may also have roles in the pathophysiology of Received: 4 June 2007, revised 29 September 2008, haemolytic and non-haemolytic transfusion reactions. accepted 16 October 2008, Key words: Chemokines, chemokine receptors, haematopoietic stem cell transplantation, published online 8 December 2008 graft-vs-host disease, transfusion reactions. General characteristics of chemokines This classification is not completely definite, as under some conditions homeostatic chemokines are inducible. Chemokines are small, secreted proteins in the range of 8– Most chemokines were originally named for their first 10 kDa that have numerous functions in normal physiology identified biological activity, such as monocyte chemoattractant and pathology. -

The Role of CCL5/RANTES in Regulating Cellular Metabolism in Activated T Cells
The Role of CCL5/RANTES in Regulating Cellular Metabolism in Activated T cells by Olivia Chan A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Immunology University of Toronto © Copyright by Olivia Chan, 2011 The Role of CCL5/RANTES in Regulating Cellular Metabolism in Activated T cells Olivia Chan Master of Science Graduate Department of Immunology University of Toronto, 2011 Recruitment of effector T cells to sites of infection is essential for an effective adaptive immune response. The inflammatory chemokine CCL5/RANTES activates its cognate receptor, CCR5, to initiate cellular functions including chemotaxis. This thesis describes the signaling events invoked by CCL5 and its ability to regulate the energy status of activated T cells. CCL5 treatment in ex vivo activated human T cells induced the activation of AMPK and downstream substrates ACC1, PFKFB2 and GSK-3. Evidence is provided that CCL5 treatment is able to induce glucose uptake in an mTOR-dependent manner. Using 2-deoxy-D-glucose, an inhibitor of glucose uptake, and Compound C, an inhibitor of AMPK, evidence is provided that demonstrate that CCL5-mediated chemotaxis is dependent on metabolic events, since these inhibitors perturb chemotaxis in a dose-dependent manner. Collectively, these studies suggest that CCL5 may also influence the metabolic status of activated T cells by simultaneously activating the AMPK and mTOR pathways. ii ACKNOWLEDGEMENTS I would like to dedicate this thesis to everyone who supported me throughout my graduate studies, and would like to express my gratitude to the individuals whom, without which, none of this could have been possible. -

Development of PY159, a Monoclonal Antibody That Repolarizes Tumor
Development of PY159, a monoclonal antibody that repolarizes tumor-associated inhibitory myeloid cells, for the treatment of solid tumors 2 Tower Place, Suite 800, Linda Liang, Chris Chan, Venkataraman Sriram, Erick Lu, Joshua L. Pollack, Mikhail Binnewies, Xiaoyan Du, Aritra Pal, Vladi Juric, South San Francisco, CA 94080 Nadine Jahchan, Evan Greger, Kevin P. Baker, Len Reyno, and Michel Streuli Introduction TREM1 Expression is Higher in Diverse Cancers, and Inversely PY159m Induces Anti-tumor Immunity Correlates With Patient Survival Dosed Dosed Dosed MethodsDosed The major differentially expressed “pathway families” in To improve the proportion of patients who benefit from checkpoint inhibitor (CPI) therapy 1500 PY159m treated MC38 tumors MC38 mono-therapy • Immune pathways Strata + trem1median=TREM1hi + trem1median=TREM1low ) • Calcium ion transport additional immune pathways likely need to be targeted. Tumor-associated macrophages TREM1 mRNA expression in 3 Negative correlation between TREM1 1000 • Tissue remodeling colorectal cancer • Metabolism and RNA processing 1.00 ++ expression and patient survival also seen in: (TAMs), myeloid-derived suppressor cells (MDSCs), and tumor-associated neutrophils (TANs) ++++++ ++ ++++++ ++ + 500 +++ + exhibit a spectrum of functional phenotypes ranging from immunosuppressive M2-like +++++ • Breast cancer Tumor volume (mm Control The immune pathway families ++ ++ 6 6 ++++++++ +++++++ PY159m + + • Antigen processing and presentation • IFN-g production + +++ • Pancreatic cancer macrophages or N2-like neutrophils that promote tumor growth to pro-inflammatory M1-like 0.75 ++ +++++ 0 • T Cell Receptor signaling pathway • TNF superfamily cytokine production + +++++++++++++ 9 15 18 21 24 27 ++++ +++++++++ • Response to TNF • Regulation of IL6 production ++ +++ Day post implant • NF KB signaling • Regulation of secretion ++++ +++++++++++++ Low • SCC ++++++ +++ TREM1 macrophages and N1-like neutrophils that promote anti-tumor immunity. -

Epithelial–Immune Cell Interplay in Primary Sjögren Syndrome Salivary Gland Pathogenesis
REVIEWS Epithelial–immune cell interplay in primary Sjögren syndrome salivary gland pathogenesis Gwenny M. Verstappen1,2, Sarah Pringle 1,2, Hendrika Bootsma1 ✉ and Frans G. M. Kroese1 Abstract | In primary Sjögren syndrome (pSS), the function of the salivary glands is often considerably reduced. Multiple innate immune pathways are likely dysregulated in the salivary gland epithelium in pSS, including the nuclear factor-κB pathway, the inflammasome and interferon signalling. The ductal cells of the salivary gland in pSS are characteristically surrounded by a CD4+ T cell-rich and B cell-rich infiltrate, implying a degree of communication between epithelial cells and immune cells. B cell infiltrates within the ducts can initiate the development of lymphoepithelial lesions, including basal ductal cell hyperplasia. Vice versa, the epithelium provides chronic activation signals to the glandular B cell fraction. This continuous stimulation might ultimately drive the development of mucosa-associated lymphoid tissue lymphoma. This Review discusses changes in the cells of the salivary gland epithelium in pSS (including acinar, ductal and progenitor cells), and the proposed interplay of these cells with environmental stimuli and the immune system. Current therapeutic options are insufficient to address both lymphocytic infiltration and salivary gland dysfunction. Successful rescue of salivary gland function in pSS will probably demand a multimodal therapeutic approach and an appreciation of the complicity of the salivary gland epithelium in the development of pSS. Nearly 100 years ago, Henrik Sjögren described a dis striated ducts, consist mostly of CD4+ T cells and B cells, ease whereby patients had a reduced functionality of although other immune cells, including (but not lim the salivary glands (xerostomia) and lacrimal glands ited to) myeloid dendritic cells, plasmacytoid dendritic (xerophthalmia), with accompanying disturbances of the cells (pDCs) and follicular dendritic cells, might also cornea and the conjunctiva (keratoconjunctivitis sicca)1. -
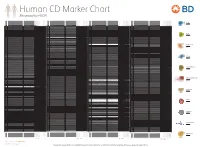
Reviewed by HLDA1
Human CD Marker Chart Reviewed by HLDA1 T Cell Key Markers CD3 CD4 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD8 CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD74 DHLAG, HLADG, Ia-g, li, invariant chain HLA-DR, CD44 + + + + + + CD158g KIR2DS5 + + CD248 TEM1, Endosialin, CD164L1, MGC119478, MGC119479 Collagen I/IV Fibronectin + ST6GAL1, MGC48859, SIAT1, ST6GALL, ST6N, ST6 b-Galactosamide a-2,6-sialyl- CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD75 CD22 CD158h KIR2DS1, p50.1 HLA-C + + CD249 APA, gp160, EAP, ENPEP + + tranferase, Sialo-masked lactosamine, Carbohydrate of a2,6 sialyltransferase + – – + – – + – – CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD75S a2,6 Sialylated lactosamine CD22 (proposed) + + – – + + – + + + CD158i KIR2DS4, p50.3 HLA-C + – + CD252 TNFSF4, -

Chemokine and Chemokine Receptor Profiles in Metastatic Salivary Adenoid Cystic Carcinoma ASHLEY C
ANTICANCER RESEARCH 36 : 4013-4018 (2016) Chemokine and Chemokine Receptor Profiles in Metastatic Salivary Adenoid Cystic Carcinoma ASHLEY C. MAYS, XIN FENG, JAMES D. BROWNE and CHRISTOPHER A. SULLIVAN Department of Otolaryngology, Wake Forest School of Medicine, Winston Salem, NC, U.S.A. Abstract. Aim: To characterize the chemokine pattern in distant metastasis (2-5). According to Ko et al. , 75% of metastatic salivary adenoid cystic carcinoma (SACC). patients with initial nodal involvement eventually developed Materials and Methods: Real-time polymerase chain distant metastasis. Patients with lung metastasis have a poor reaction (RT-PCR) was used to compare chemokine and prognosis (6). chemokine receptor gene expression in two SACC cell lines: The development of distant metastatic disease is the chief SACC-83 and SACC-LM (lung metastasis). Chemokines and cause for mortality (7, 8). Primary treatment is complete receptor genes were then screened and their expression surgical resection when feasible with adjuvant radiotherapy. pattern characterized in human tissue samples of non- The role of chemotherapy is debatable. Treatment of recurrent SACC and recurrent SACC with perineural metastatic ACC has been difficult to date due to lack of invasion. Results: Expression of chemokine receptors specific targets for metastatic cells (1). Though the steps that C5AR1, CCR1, CCR3, CCR6, CCR7, CCR9, CCR10, must occur in the metastatic event are well characterized, it CXCR4, CXCR6, CXCR7, CCRL1 and CCRL2 were higher remains unclear why or how ACC cells ultimately “choose” in SACC-83 compared to SACC-LM. CCRL1, CCBP2, or are ”chosen” to migrate to a specific metastatic site. A CMKLR1, XCR1 and CXCR2 and 6 chemokine genes mounting body of evidence suggests that cytokine-like (CCL13, CCL27, CXCL14, CMTM1, CMTM2, CKLF) were molecules called chemokines play a significant role in more highly expressed in tissues of patients without tumor directing the cellular traffic in metastatic melanoma, lung, recurrence/perineural invasion compared to those with breast and ACC cancers (9-15). -

CCL26 Regulates the Proportion of CD4+CD25+FOXP3+ Tregs and The
EXPERIMENTAL AND THERAPEUTIC MEDICINE 20: 3343-3351, 2020 CCL26 regulates the proportion of CD4+CD25+FOXP3+ Tregs and the production of inflammatory factors in peripheral blood mononuclear cells following acute ischemic stroke via the STAT5 pathway ZHIQIANG DONG1*, LIMEI CAO1*, LAN GUO2, YUAN HONG2, JINXIU CAO2 and XU CHEN1 1Department of Neurology, Shanghai Eighth People's Hospital; 2Geriatric Nursing Ward, Shanghai Eighth People's Hospital, Shanghai 200233, P.R. China Received January 20, 2020; Accepted May 20, 2020 DOI: 10.3892/etm.2020.9046 Abstract. Acute ischemic stroke (AIS) is the most common of inflammatory factors in PBMCs following AIS via the type of stroke. Recent studies have found that AIS is closely STAT5 pathway. involved in the immune regulation function of regulatory T cells (Tregs). C-C motif chemokine ligand 26 (CCL26) is a Introduction member of the chemokine family that plays an essential role in cell activation, cell differentiation, lymphocyte homing, At present, stroke has become the second most leading cause and inflammatory and immune responses. The present study of mortality worldwide and is also one of the chief factors aimed to investigate the role of CCL26 in the regulation of causing long-term disability among middle-aged and older Tregs in AIS. Peripheral blood mononuclear cells (PBMCs) individuals (1,2). There are approximately 17 million newly were incubated with a CCL26-neutralizing antibody. The diagnosed stroke cases each year worldwide. In Western proportion of cluster of differentiation (CD)4+CD25+ forkhead countries, stroke is the most common lethal disease ranked box P3 (FOXP3)+ Tregs was increased, and the expression of after cancer and acute myocardial infarction, with acute isch- FOXP3, phosphorylated signal transducer and activator of emic stroke (AIS) accounting for approximately 80% of all transcription 5 (p-STAT5), and that of the immunosuppressive stroke cases (3,4).