Compound 1080), Its Pharmacology and Toxicology Ernest Kun
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Basic Aspects of Fluorine in Chemistry and Biology
Introduction: Basic Aspects of Fluorine in Chemistry and Biology COPYRIGHTED MATERIAL 1 Unique Properties of Fluorine and Their Relevance to Medicinal Chemistry and Chemical Biology Takashi Yamazaki , Takeo Taguchi , and Iwao Ojima 1.1 Fluorine - Substituent Effects on the Chemical, Physical and Pharmacological Properties of Biologically Active Compounds The natural abundance of fl uorine as fl uorite, fl uoroapatite, and cryolite is considered to be at the same level as that of nitrogen on the basis of the Clarke number of 0.03. However, only 12 organic compounds possessing this special atom have been found in nature to date (see Figure 1.1) [1] . Moreover, this number goes down to just fi ve different types of com- pounds when taking into account that eight ω - fl uorinated fatty acids are from the same plant [1] . [Note: Although it was claimed that naturally occurring fl uoroacetone was trapped as its 2,4 - dinitrohydrazone, it is very likely that this compound was fl uoroacetal- dehyde derived from fl uoroacetic acid [1] . Thus, fl uoroacetone is not included here.] In spite of such scarcity, enormous numbers of synthetic fl uorine - containing com- pounds have been widely used in a variety of fi elds because the incorporation of fl uorine atom(s) or fl uorinated group(s) often furnishes molecules with quite unique properties that cannot be attained using any other element. Two of the most notable examples in the fi eld of medicinal chemistry are 9α - fl uorohydrocortisone (an anti - infl ammatory drug) [2] and 5 - fl uorouracil (an anticancer drug) [3], discovered and developed in 1950s, in which the introduction of just a single fl uorine atom to the corresponding natural products brought about remarkable pharmacological properties. -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

Enzymes Quinate Dehydrogenase
THE ABSOLUTTE STEREOCHEMICAL COUtRSE OF CITRIC ACID BIOSYNTHESIS* BY KENNETH R. HANSON AND IRWIN A. RoSEt DEPARTMENTS OF BIOCHEMISTRY OF THE CONNECTICUT AGRICULTURAL EXPERIMENT STATION AND OF YALE UNIVERSITY SCHOOL OF MEDICINE, NEW HAVEN Communicated by H. B. Vickery, September 3, 1963 In 1948 Ogston noted that when such symmetrical molecules as citric (or amino- malonic) acid are acted upon by asymmetric enzyme surfaces, pairs of identical groups may well be differentiated.1 Shortly thereafter it was shown, by using labeled substrates, that the two -CH2COOH groups of citric acid could, in fact, be distinguished.2 When citrate is formed from oxaloacetate and acetyl-CoA by citrate synthase (E.C.4.1.3.7, condensing enzyme), and is then treated with aco- nitate hydratase (E.C.4.2.1.3, aconitase) to form cis-aconitate and threo-Dr-iso- citrate, the -CH2COOH ends of these compounds are derived from acetyl-CoA, while the other atoms are derived either from oxaloacetate or water.3-6 The acetyl-CoA-derived group yields acetate when citrate is cleaved by citrate-lyase (E.C.4.1.3.6, citratase),7'8 and acetyl-CoA when citrate is attacked by the citrate cleavage enzyme.9 The nonequivalence of the two -CH2COOH groups is apparent when citric acid is represented as I (heavy line above, dotted lines below the plane of the paper). Carbon atoms 1 and 2 are to 5 and 4 as right arm is to left arm:'0 they cannot be superimposed except by reflection. The same molecule is shown in Emil Fischer projection as II (vertical lines below and horizontal lines above the plane of the paper). -

Long-Chain F-18 Fatty Acids for the Study of Regional Metabolism in Heart and Liver; Odd-Even Effects of Metabolism in Mice
BASIC SCIENCES RADIOCHEMISTRY AND RADIOPHARMACEUTICALS Long-Chain F-18 Fatty Acids for the Study of Regional Metabolism in Heart and Liver; Odd-Even Effects of Metabolism in Mice E. J. Knust, Ch. Kupfernagel, and G. Stöcklin Institut fur Chemie 1 (Nuklearchemie) der KFA Julich GmbH, D-5170 Julich, FRG ( West Germany) In view of the potential usefulness of fluorine-tagged fatty acids in the study of regional metabolism in the heart and liver, the time courses of uptake and release of 9,10-[18F)fluorostearic acid, 2-{18F]fluorostearic acid, 16-{18F]fluorohexadec- anoic acid, 17-[18F]fluoroheptadecanoic acid have been investigated in several organs of NMRI mice. Whereas 2-[18F]fluorostearic acid shows very little uptake in the heart muscle but an increasing accumulation In the fiver, the fatty acids with the F-18 label in the middle or at the end of the carbon chain exhibit uptake and elimination behavior similar to that of the analogous C-11 -labeled compounds. After rapid concentration in the heart within 1 min, clearance takes place with fast and slow components. 16-| 18F|fluorohexadecanoic acid and 17-|18F|fluorohepta- decanoic acid have different half-times of elimination. These differences are also reflected by the fact that nearly all the activity present in the heart can be recov ered as fluoride(F-18) in the case of 17-|18F|fluoroheptadecanoic acid, whereas practically no fluoride was found among the metabolites of 16-[18F]fluorohexadec- anoic acid. Similar differences were observed for the F-18 activity in bone. The results can be interpreted in terms of the odd-even rule: ßoxidation of even-num bered fatty acids ends up with 118F|fluoroacetic acid, whereas the odd-numbered fatty acids give rise to ß-|18F|fluoropropionic acid. -

Toxic Characteristics of Fluorocitrate, the Toxic Metabolite of Compound 1080 Peter J, Savarje
TOXIC CHARACTERISTICS OF FLUOROCITRATE, THE TOXIC METABOLITE OF COMPOUND 1080 PETER J, SAVARJE. Denver Wildlife Research Center. U.S. Flsh and Wllclllfe Service, Building 16, Federal Center. Denver. Colorado 80225 ABSTRACT: This paper reviews toxicological research involving fluorocitrate, the toxic metabolite of sodium monofluoroacetate (fluoroacetate), which is the active ingredient in the pesticide Compound 1080. Many toxicological studies have been done with fluoroacetate and the results obtained are actually due to the fluorocitrate because it has been definitely proved that, from a biochemical perspective,fluoro acetate is not toxic but fluorocitrate is. The classical explanation of the toxic action of fluoroci trate is that it inhibits the enzyme aconitase in the tricarboxylic acid cycle. Deactivation of aconitase results in decreased energy production by cells and ultimately death of the organism. However, the more recent explanation of fluorocitrate's mode of action is that it binds with mito chondrial protein which prevents transport of citrate and its utilization by cells for energy production. Metabolism ~tudies indicate that only small amounts, perhaps less than 3%, of fluorocitrate is fonned from fluoroacetate. From the limited number of acute and chronic studies conducted with fluorocitrate it does not appear to be as potent as fluoroacetate by either the oral or parenteral routes of admini· stration. This decreased level of toxicity is thought to be due to the larger molecular weight of fluorocitrate which would not be as readily absorbed by tissues. Central nervous system toxic mani festations (i.e., tremors, convulsions) are characteristic in many animals poisoned with fluoroacetate. Fluorocitrate administered directly into the brain was found to be 100 times more toxic than fluoro acetate. -

HRE05002-004.Pdf(PDF, 1.7
1080 Reassessment Application October 2006 Appendix C Source: Landcare Research (1964). Control of poisons. Royal Society of Health Journal 84, 52-53. Keywords: poisons/non-target species/fluoroacetamide/livestock Occupational Health Bulletin: Sodium Fluoroacetate Compound 1080. New Series No 1 (revision of Vol.6 No 11, July 1962). 1967. Wellington, Department of Health. Ref Type: Pamphlet Keywords: sodium fluoroacetate/fluoroacetate/1080 (1969). Fluoroacetate. In 'Clinical toxicology of commercial products'. (M. Gleason, R. Gosselin, H. Hodge, and R. SmithEds. ) pp. 116-117. (The Williams & Wilkins: Baltimore.) Keywords: fluoroacetate/sodium fluoroacetate/diagnosis/treatment/acute toxicity Poisonings. 20. 1976. Surveillance 1976 No.4. Ref Type: Report Keywords: poisoning/1080/analysis/muscle/liver/livestock/witholding period Abstract: 1080 poisoning was in the public eye in Canterbury when sheep died after they were returned to a block pronounced "safe" after poisoning operations. About 160 ewes died out of 800, and 1080 poisoning was confirmed. It is reported that errors were made in the analysis of bait tested to determine if it was safe to stock. Recently a workshop on 1080 analysis was held at Invermay AHL. These are the recommendations for sampling: 1) Take the samples from the animals which are first to die in the outbreak even though they may be more autolysed. 2) The best specimens in order of preference are muscle, stomach contents then liver 1080 poisoning. 26. 1976. Surveillance 1976 No. 4. Ref Type: Report Keywords: 1080/poisoning/birds/persistence in animals/non-target species/secondary poisoning/humans Abstract: Recently, Canada geese around Lake Benmore were poisoned by oats impregnated with 1080 Diagnosis of 1080 poisoning in dogs. -

Fluoroacetate Resistance in Streptomyces Cattleya 3.1 Introduction 41 3.2 Materials and Methods 42 3.3 Results and Discussion 47 3.4 Conclusions 66 3.5 References 67
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Expanding the scope of organofluorine biochemistry through the study of natural and engineered systems Permalink https://escholarship.org/uc/item/9n89c8zv Author Walker, Mark Chalfant Publication Date 2013 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Expanding the scope of organofluorine biochemistry through the study of natural and engineered systems by Mark Chalfant Walker A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Molecular and Cell Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Michelle C. Y. Chang, Chair Professor Judith P. Klinman Professor Susan Marqusee Professor Mathew B. Francis Spring 2013 Expanding the scope of organofluorine biochemistry through the study of natural and engineered systems © 2013 by Mark Chalfant Walker Abstract Expanding the scope of organofluorine biochemistry through the study of natural and engineered systems by Mark Chalfant Walker Doctor of Philosophy in Molecular and Cell Biology University of California, Berkeley Professor Michelle C. Y. Chang, Chair Fluorination has become a very useful tool in the design and optimization of bioactive small molecules ranging from pesticides to pharmaceuticals. Its small size allows a sterically conservative substitution for a hydrogen or hydroxyl, thus maintaining the overall size and shape of a molecule. However, the extreme electronegativity of fluorine can dramatically alter other properties of the molecule. As a result, the development of new methods for fluorine incorporation is currently a major focus in synthetic chemistry. It is our goal to use a complementary biosynthetic approach to use enzymes for the regio-selective incorporation of fluorine into complex natural product scaffolds through the fluoroacetate building block. -

Accelerated Discovery Chemistry Using Fluoroorganic Ligands
Accelerated Discovery Chemistry Using Fluoroorganic Ligands 228th ACS National Meeting Philadelphia, PA Andrei A. Gakh, PhD Oak Ridge National Laboratory August 22, 2004 OAK RIDGE NATIONAL LABORATORY U. S. DEPARTMENT OF ENERGY 1 Discovery Chemistry Project an Overview • Synthesis and utilization of novel organic compounds by Objective U.S. National Laboratories, chemical companies, and other government and non-government organizations. • An international project offering the combination of Approach expertise and resources of the participants in a team effort under the guidance and financial support of the Initiatives for Proliferation Prevention. OAK RIDGE NATIONAL LABORATORY U. S. DEPARTMENT OF ENERGY 2 Project-at-Glance Resources Industry National Labs Universities International Resources: • Industry: 52% • National Laboratories: 13% • Universities: 4% • International Collaborators: 31% Major USG Funding: • US DOE, National Nuclear Security Administration •Initiatives for Proliferation Prevention OAK RIDGE NATIONAL LABORATORY U. S. DEPARTMENT OF ENERGY 3 Major Research Directions Information Synthesis Management Banks of Compounds System More than 60% of the resources are allocated to biological targets (biological discovery) OAK RIDGE NATIONAL LABORATORY U. S. DEPARTMENT OF ENERGY 4 Biological Targets The World of Big Numbers and Low Yields Recent example: • 39,647 structures proposed •11,248 structures accepted •6,217 compounds prepared •5,981 passed quality control •5,624 screened •127 active compounds identified •32 active leads selected Yield: 0.081% (active leads) Human cost: >70 FTE (or > 2 FTE to develop an active lead) OAK RIDGE NATIONAL LABORATORY U. S. DEPARTMENT OF ENERGY 5 Complexity of Biological Targets GABA Receptors The GABAA receptor is a pentamer consisting of various combinations of alpha, beta, and gamma subunits. -
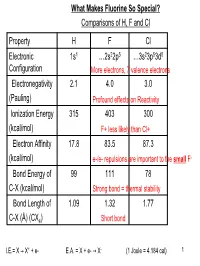
What Makes Fluorine So Special? Comparisons of H, F and Cl Property H F Cl Electronic Configuration 1S1 …2S22p5 …3S23p53d0 E
What Makes Fluorine So Special? Comparisons of H, F and Cl Property H F Cl Electronic 1s1 …2s22p5 …3s23p53d0 Configuration More electrons, 7 valence electrons Electronegativity 2.1 4.0 3.0 (Pauling) Profound effects on Reactivity Ionization Energy 315 403 300 (kcal/mol) F+ less likely than Cl+ Electron Affinity 17.8 83.5 87.3 (kcal/mol) e-/e- repulsions are important to the small F- Bond Energy of 99 111 78 C-X (kcal/mol) Strong bond = thermal stability Bond Length of 1.09 1.32 1.77 C-X (Å) (CX4) Short bond I.E.= X → X+ + e- E.A. = X + e- → X- (1 Joule = 4.184 cal) 1 Polythenes: H2 F2 Cl2 C C C C n C n C n H2 F2 Cl2 stable stable unstable (Shows similarity between H and F – actually the STERICS make the Cl version unstable) Preference as Leaving Groups H+ F- Cl- (Shows difference between H and F. This is an Electronics issue) 2 1H and 19F NMR Nucleus 1H 19F Rel. Sensitivity 1.00 0.834 Spin 1/2 1/2 Abundance 99.98% 100% NMR Frequency in 42.576 40.055 MHz for 1T field Typical δ range /ppm 10ppm 260ppm larger spectral width means spectra are more informative 3 Bond Energies for Halogens (in kcal/mol) Halogen X-X H-X BX3 AlX3 CX4 F 37.7 136.0 154.2 139.1 109.0 Cl 58.2 103.0 106.1 102.1 78.2 Br 46.1 88.0 88.0 86.0 65.0 I 36.1 71.0 65.0 68.1 57.1 The F-F bond is weak, too many electrons in a confined area. -

Carbon-Fluorine Compounds Chemistry, Biochemistry & Biological Activities
Carbon-Fluorine Compounds Chemistry, Biochemistry & Biological Activities A Ciba Foundation Symposium 1972 Elsevier - Excerpta Medica North-Holland Associated Scientific Publishers . Amsterdam - London . New York Carbon-Fluorine Compounds Chemistry, Biochemistry & Biological Activities The Ciba Foundation for the promotion of international cooperation in medical and chemical research is a scientific and educational charity established by CIBA Limited - now CIBA-GEIGYLimited - of Basle. The Foundation operates independently in London under English trust law. Ciba Foundation Symposia are published in collaboration with Associated Scientific Publishers (Elsevier Scientific Publishing Company, Excerpta Medica, North-Holland Publishing Company) in Amsterdam. Associated Scientific Publishers, P.O. Box 3489, Amsterdam Carbon-Fluorine Compounds Chemistry, Biochemistry & Biological Activities A Ciba Foundation Symposium 1972 Elsevier - Excerpta Medica North-Holland Associated Scientific Publishers . Amsterdam - London . New York 0 Copyright 1972 Ciba Foundation All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying and recording, or by any information storage and retrieval system, without permission in writing from the publishers. ISBN Excerpta Media 90 219 4002 7 ISBN American Elsevier 0-444-10373-2 Library of Congress Catalog Card Number 72-76005 Published in 1972 by Associated Scientific Publishers, P.O. Box 3489, Amsterdam, and 52 Vanderbilt Avenue, New York, N.Y. 10017. Suggested series entry for library catalogues: Ciba Foundation Symposia. Printed in The Netherlands by Mouton & Co., The Hague Contents SIR RUDOLPH PETERS Introduction 1 B. c. SAUNDERS Chemical characteristics of the carbon-fluorine bond 9 Discussion 27 A. G. SHARPE The physical properties of the carbon-fluorine bond 33 Discussion 49 SIR RUDOLPH PETERS Some metabolic aspects of fluoroacetate especially related to fluorocitrate 55 Discussion 70 E. -

Toxicology of Fluoroacetate: a Review, with Possible Directions for Therapy
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/7515021 Toxicology of fluoroacetate: A review, with possible directions for therapy research Article in Journal of Applied Toxicology · March 2006 DOI: 10.1002/jat.1118 · Source: PubMed CITATIONS READS 88 3,015 3 authors, including: Nikolay V Goncharov 109 PUBLICATIONS 660 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: Analysis of the chemical composition of the products of La-catalyzed mathanolysis of Russian VX View project The Effects of Weakened Geomagnetic Field View project All content following this page was uploaded by Nikolay V Goncharov on 25 January 2018. The user has requested enhancement of the downloaded file. 148JOURNALN. V. GONCHAROVOF APPLIED TOXICOLOGYET AL. J. Appl. Toxicol. 2006; 26: 148–161 Published online 26 October 2005 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/jat.1118 Toxicology of fluoroacetate: a review, with possible directions for therapy research Nikolay V. Goncharov,1 Richard O. Jenkins2,* and Andrey S. Radilov1 1 Research Institute of Hygiene, Occupational Pathology and Human Ecology, Saint-Petersburg, Russia 2 School of Allied Health Sciences, De Montfort University, Leicester, UK Received 30 June 2005; Revised 16 August 2005; Accepted 5 September 2005 ABSTRACT: Fluoroacetate (FA; CH2FCOOR) is highly toxic towards humans and other mammals through inhibition of the enzyme aconitase in the tricarboxylic acid cycle, caused by ‘lethal synthesis’ of an isomer of fluorocitrate (FC). FA is found in a range of plant species and their ingestion can cause the death of ruminant animals. -

REVIEW an Updated Review of the Toxicology and Ecotoxicology of Sodium Fluoroacetate (1080) in Relation to Its Use As a Pest Control Tool in New Zealand
AvailableEason et al.:on-line Update at: http://www.newzealandecology.org/nzje/ on 1080 in New Zealand 1 REVIEW An updated review of the toxicology and ecotoxicology of sodium fluoroacetate (1080) in relation to its use as a pest control tool in New Zealand Charles Eason1*, Aroha Miller1, Shaun Ogilvie1 and Alastair Fairweather2 1Faculty of Agriculture and Life Sciences, Department of Ecology, Lincoln University, PO Box 84, Lincoln 7647, New Zealand 2 Department of Conservation, Hamilton, New Zealand * Author for correspondence (Email: [email protected]) Published on-line: 19 December 2010 Abstract: Sodium fluoroacetate (1080) is a vertebrate pesticide, originally developed in the 1940s and principally used for the control of unwanted introduced animals in New Zealand and Australia. Fluoroacetate is also a toxic component of poisonous plants found in Australia, Africa, South America, and India. In relation to its use as a pesticide, recent research has focused on further elucidation of its potential sub-lethal effects, on animal welfare issues, on understanding and reducing its risk to non-target species, on its ecotoxicology, and fate in the environment following use in baits. 1080 acts by interfering with cellular energy production through inhibition of the tricarboxylic acid cycle and lethal doses can kill animal pests within 6–48 h of eating baits. Exposure to sub-lethal doses has been shown to have harmful effects on the heart and testes in animal studies, and strict safety precautions are enforced to protect contractors and workers in the pest control industry. Considerable care must be taken when using 1080 for the control of animal pests.