Utilization of a Helium Discharge Detector for Quantitation of Organohalogen Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis of Bromochloromethane Using Phase Transfer Catalysis
1 SYNTHESIS OF BROMOCHLOROMETHANE USING PHASE TRANSFER CATALYSIS By LANCELOT LUCRETIUS BROOKS Baccalaureus Scientiae Honores-Chemistry, Nelson Mandela Metropolitan University A dissertation submitted in fulfillment of the requirements for the Masters Degree in Chemistry In the Faculty of Science at the NELSON MANDELA METROPOLITAN UNIVERSITY Nov. 2011 Promoter : Dr G. Dugmore Co-Promoter : Prof B. Zeelie 2 DECLARATION I, Lancelot Brooks, hereby declare that the above-mentioned treatise is my own work and that it has not previously been submitted for assessment to another University, or for another qualification. ……………………………….. ……………………….. Mr. L.L. Brooks Date 3 ACKNOWLEDGEMENTS To my promoters Dr. Gary Dugmore, and Prof. Ben Zeelie for their invaluable input, help and guidance. To NRF and NMMU for financial assistance To my parents and brothers for their love and support To Peter, Batsho, Unati, and friends in NMMU chemistry research laboratory, thank you guys. To my dearest fiancée, Natasha, a very special thank you for always being there and supporting me. Love you angel. “And we know that all things work together for good to those who love God, to those who are called according to His purpose” -Romans 8:28. 4 TABLE OF CONTENTS DECLARATION……………………………………………………………………. 2 ACKNOWLEDGEMENTS……………………………………………………………. 3 TABLE OF CONTENTS………………………………………………………………. 4 LIST OF FIGURES…………………………………………………………………….. 8 LIST OF TABLES……………………………………………………………………… 9 LIST OF EQUATIONS………………………………………………………………… 11 SUMMARY……………………………………………………………………………… 12 CHAPTER 1…………………………………………………………………………….. 14 INTRODUCTION………………………………………………………………………. 14 1.1. Technology of leather production……………………………………………….. 14 1.2. Synthesis of TCMTB……………………………………………………………… 17 1.3. Bromine……………………………………………………………………………. 20 1.3.1. Overview……………………………………………………………. 20 1.3.2. Applications of bromine compounds…………..…………………. 22 1.3.2.1. Photography……………………………………………… 22 1.3.2.2. -

Safe Handling and Disposal of Chemicals Used in the Illicit Manufacture of Drugs
Vienna International Centre, PO Box 500, 1400 Vienna, Austria Tel.: (+43-1) 26060-0, Fax: (+43-1) 26060-5866, www.unodc.org Guidelines for the Safe handling and disposal of chemicals used in the illicit manufacture of drugs United Nations publication USD 26 Printed in Austria ISBN 978-92-1-148266-9 Sales No. E.11.XI.14 ST/NAR/36/Rev.1 V.11-83777—September*1183777* 2011—300 Guidelines for the Safe handling and disposal of chemicals used in the illlicit manufacture of drugs UNITED NATIONS New York, 2011 Symbols of United Nations documents are composed of letters combined with figures. Mention of such symbols indicates a reference to a United Nations document. ST/NAR/36/Rev.1 UNITED NATIONS PUBLICATION Sales No. E.11.XI.14 ISBN 978-92-1-148266-9 eISBN 978-92-1-055160-1 © United Nations, September 2011. All rights reserved. The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. Requests for permission to reproduce this work are welcomed and should be sent to the Secretary of the Publications Board, United Nations Headquarters, New York, N.Y. 10017, U.S.A. or also see the website of the Board: https://unp.un.org/Rights.aspx. Governments and their institutions may reproduce this work without prior authoriza- tion but are requested to mention the source and inform the United Nations of such reproduction. -

Maine Remedial Action Guidelines (Rags) for Contaminated Sites
Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites (RAGs) Effective Date: May 1, 2021 Approved by: ___________________________ Date: April 27, 2021 David Burns, Director Bureau of Remediation & Waste Management Executive Summary MAINE DEPARTMENT OF ENVIRONMENTAL PROTECTION 17 State House Station | Augusta, Maine 04333-0017 www.maine.gov/dep Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites Contents 1 Disclaimer ...................................................................................................................... 1 2 Introduction and Purpose ............................................................................................... 1 2.1 Purpose ......................................................................................................................................... 1 2.2 Consistency with Superfund Risk Assessment .............................................................................. 1 2.3 When to Use RAGs and When to Develop a Site-Specific Risk Assessment ................................. 1 3 Applicability ................................................................................................................... 2 3.1 Applicable Programs & DEP Approval Process ............................................................................. 2 3.1.1 Uncontrolled Hazardous Substance Sites ............................................................................. 2 3.1.2 Voluntary Response Action Program -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2. -

Federal Register/Vol. 84, No. 230/Friday, November 29, 2019
Federal Register / Vol. 84, No. 230 / Friday, November 29, 2019 / Proposed Rules 65739 are operated by a government LIBRARY OF CONGRESS 49966 (Sept. 24, 2019). The Office overseeing a population below 50,000. solicited public comments on a broad Of the impacts we estimate accruing U.S. Copyright Office range of subjects concerning the to grantees or eligible entities, all are administration of the new blanket voluntary and related mostly to an 37 CFR Part 210 compulsory license for digital uses of increase in the number of applications [Docket No. 2019–5] musical works that was created by the prepared and submitted annually for MMA, including regulations regarding competitive grant competitions. Music Modernization Act Implementing notices of license, notices of nonblanket Therefore, we do not believe that the Regulations for the Blanket License for activity, usage reports and adjustments, proposed priorities would significantly Digital Uses and Mechanical Licensing information to be included in the impact small entities beyond the Collective: Extension of Comment mechanical licensing collective’s potential for increasing the likelihood of Period database, database usability, their applying for, and receiving, interoperability, and usage restrictions, competitive grants from the Department. AGENCY: U.S. Copyright Office, Library and the handling of confidential of Congress. information. Paperwork Reduction Act ACTION: Notification of inquiry; To ensure that members of the public The proposed priorities do not extension of comment period. have sufficient time to respond, and to contain any information collection ensure that the Office has the benefit of SUMMARY: The U.S. Copyright Office is requirements. a complete record, the Office is extending the deadline for the extending the deadline for the Intergovernmental Review: This submission of written reply comments program is subject to Executive Order submission of written reply comments in response to its September 24, 2019 to no later than 5:00 p.m. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0314758 A1 Gross0 Et Al
US 20080314758A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0314758 A1 Gross0 et al. (43) Pub. Date: Dec. 25, 2008 (54) PROCESS FOR CONVERTING Related U.S. Application Data HYDROCARBON FEEDSTOCKS WITH ELECTROLYTC RECOVERY OF HALOGEN (60) Provisional application No. 60/930,220, filed on May 14, 2007. (75) Inventors: Philip Grosso, Auburn, CA (US); Publication Classification Eric W. McFarland, Santa Barbara, (51) Int. Cl CA (US); Jeffrey H. Sherman, we WE FL C25B 3/06 (2006.01) s C25B 3/00 (2006.01) Correspondence Address: (52) U.S. Cl. .......... 205/431; 205/462; 205/451; 204/277 CHRISTIE, PARKER & HALE, LLP (57) ABSTRACT PO BOX 7O68 PASADENA, CA 91109–7068 (US) An improved continuous process for converting methane, natural gas, and other hydrocarbon feedstocks into one or more higher hydrocarbons, methanol, amines, or other prod (73) Assignee: GRT, INC. ucts comprises continuously cycling through hydrocarbon halogenation, product formation, product separation, and (21) Appl. No.: 12/152,515 electrolytic regeneration of halogen, optionally using an improved electrolytic cell equipped with an oxygen depolar (22) Filed: May 14, 2008 ized cathode. NATURAL GAS w RECYCLE & WENT 24 WET BROMINE HYDROGEN BROMINATION --2O 18-- - -- 17 ELECTROLYSIs s 19 COOLER CELS C ACID/BASE FOR pH CONTROL AQUEOUS NaBr A. AQUEOUS NaOH/NaBr ORGANICS 15- -13 14 HBr ABSORPTION NaBr- ORGANICS Aoys & 12- PRODUCT RECOVERY SEPARATION NCS PRODUCT Patent Application Publication Dec. 25, 2008 Sheet 1 of 11 US 2008/0314758A1 © Patent Application Publication Dec. 25, 2008 Sheet 3 of 11 US 2008/0314758A1 IyyanLy?ISwºTV80LVNSºº Patent Application Publication Dec. -

Risk-Based Closure Guide Draft
Risk-based Closure Guide July 1, 2021 Office of Land Quality Indiana Department of Environmental Management Disclaimer This Nonrule Policy Document (NPD) is being established by the Indiana Department of Environmental Management (IDEM) consistent with its authority under IC 13-14-1-11.5. It is intended solely as guidance and shall be used in conjunction with applicable rules or laws. It does not replace applicable rules or laws, and if it conflicts with these rules or laws, the rules or laws shall control. Pursuant to IC 13-14-1-11.5, this NPD will be available for public inspection for at least forty-five (45) days prior to presentation to the appropriate State Environmental Board, and may be put into effect by IDEM thirty (30) days afterward. If the NPD is presented to more than one board, it will be effective thirty (30) days after presentation to the last State Environmental Board. IDEM also will submit the NPD to the Indiana Register for publication. 2 Table of Contents 1 Introduction ..................................................................................................................................... 7 1.1 Applicability .............................................................................................................................. 8 1.2 Types of Closure ...................................................................................................................... 9 1.3 Process Overview ................................................................................................................... -

Defining Potential Chemical Peaks and Management Options
PROJECT NO. 4991 Defining Potential Chemical Peaks and Management Options Defining Potential Chemical Peaks and Management Options Prepared by: Jean Debroux Kennedy Jenks Consultants Megan H. Plumlee Orange County Water District Shane Trussell Trussell Technologies, Inc. Co-sponsored by: California State Water Resources Control Board 2021 The Water Research Foundation (WRF) is a nonprofit (501c3) organization which provides a unified source for One Water research and a strong presence in relationships with partner organizations, government and regulatory agencies, and Congress. The foundation conducts research in all areas of drinking water, wastewater, stormwater, and water reuse. The Water Research Foundation’s research portfolio is valued at over $700 million. WRF plays an important role in the translation and dissemination of applied research, technology demonstration, and education, through creation of research-based educational tools and technology exchange opportunities. WRF serves as a leader and model for collaboration across the water industry and its materials are used to inform policymakers and the public on the science, economic value, and environmental benefits of using and recovering resources found in water, as well as the feasibility of implementing new technologies. For more information, contact: The Water Research Foundation 1199 North Fairfax Street, Suite 900 6666 West Quincy Avenue Alexandria, VA 22314-1445 Denver, Colorado 80235-3098 www.waterrf.org P 571.384.2100 P 303.347.6100 [email protected] ©Copyright 2021 by The Water Research Foundation. All rights reserved. Permission to copy must be obtained from The Water Research Foundation. WRF ISBN: 978-1-60573-555-9 WRF Project Number: 4991 This report was prepared by the organization(s) named below as an account of work sponsored by The Water Research Foundation. -

Intermolecular Interaction in Methylene Halide (CH2F2, Ch2cl2, Ch2br2 and CH2I2) Dimers
molecules Article Intermolecular Interaction in Methylene Halide (CH2F2, CH2Cl2, CH2Br2 and CH2I2) Dimers László Almásy 1,2,* ID and Attila Bende 3,* ID 1 State Key Laboratory of Environment-friendly Energy Materials, Southwest University of Science and Technology, Mianyang 621010, China 2 Institute for Solid State Physics and Optics, Wigner Research Centre for Physics, Konkoly Thege út 29-33, 1121 Budapest, Hungary 3 Molecular and Biomolecular Physics Department, National Institute for Research and Development of Isotopic and Molecular Technologies, Donat Street, No. 67-103, Ro-400293 Cluj-Napoca, Romania * Correspondence: [email protected] (L.A.); [email protected] (A.B.) Academic Editors: Igor Reva and Xuefeng Wang Received: 27 April 2019; Accepted: 1 May 2019; Published: 10 May 2019 Abstract: The intermolecular interaction in difluoromethane, dichloromethane, dibromomethane, and diiodomethane dimers has been investigated using high level quantum chemical methods. The potential energy curve of intermolecular interaction along the C··· C bond distance obtained using the coupled-cluster theory with singles, doubles, and perturbative triples excitations CCSD(T) were compared with values given by the same method, but applying the local (LCCSD(T)) and the explicitly correlated (CCSD(T)-F12) approximations. The accuracy of other theoretical methods—Hartree–Fock (HF), second order Møller–Plesset perturbation (MP2), and dispersion corrected DFT theory—were also presented. In the case of MP2 level, the canonical and the local-correlation cases combined with the density-fitting technique (DF-LMP2)theories were considered, while for the dispersion-corrected DFT, the empirically-corrected BLYP-D and the M06-2Xexchange-correlation functionals were applied. In all cases, the aug-cc-pVTZ basis set was used, and the results were corrected for the basis set superposition error (BSSE) using the counterpoise method. -

Ethylene Dibromide
EPA Document# EPA-R-20-012 August 2020 United States Office of Chemical Safety and Environmental Protection Agency Pollution Prevention Final Scope of the Risk Evaluation for Ethylene Dibromide CASRN 106-93-4 August 2020 TABLE OF CONTENTS ACKNOWLEDGEMENTS ......................................................................................................................6 ABBREVIATIONS AND ACRONYMS ..................................................................................................7 EXECUTIVE SUMMARY .....................................................................................................................10 1 INTRODUCTION ............................................................................................................................13 2 SCOPE OF THE EVALUATION ...................................................................................................13 2.1 Reasonably Available Information..............................................................................................13 Search of Gray Literature ...................................................................................................... 14 Search of Literature from Publicly Available Databases (Peer-Reviewed Literature) .......... 15 Search of TSCA Submissions ................................................................................................ 24 2.2 Conditions of Use ........................................................................................................................25 Categories and Subcategories -
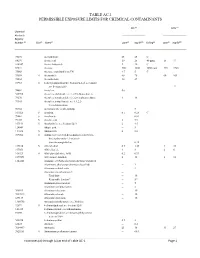
Table Ac-1 Permissible Exposure Limits for Chemical Contaminants
TABLE AC-1 PERMISSIBLE EXPOSURE LIMITS FOR CHEMICAL CONTAMINANTS PEL (d) STEL (o) Chemical Abstracts Registry Number (a) (b) (c) (e) 3(f) (g) (e) 3(f) Skin Name ppm mg/M Ceiling ppm mg/M 75070 Acetaldehyde 25 45 C 64197 Acetic acid 10 25 40 ppm 15 37 108247 Acetic Anhydride 5 20 C 67641 Acetone 500 1200 3000 ppm 750 1780 75868 Acetone cyanohydrin as CN 4.7 5 C 75058 S Acetonitrile 40 70 60 105 98862 Acetophenone 10 49 53963 S 2-Acetylaminofluorene; N-fluoren-2-yl acetamide; see Section 5209 12 74862 Acetylene (h) 540590 Acetylene dichloride; see 1,2-Dichloroethylene 79276 Acetylene tetrabromide:1,1,2,2-tetrabromoethane 1 14 79345 Acetylene tetrachloride; see 1,1,2,2- Tetrachloroethane 50782 Acetylsalicylic acid (Aspirin) 5 107028 S Acrolein 0.1 0.25 C 79061 S Acrylamide -- 0.03 79107 S Acrylic acid 2 5.9 107131 S Acrylonitrile; see Section 5213 2 4.5 124049 Adipic acid -- 5 111693 S Adiponitrile 2 8.8 309002 S Aldrin; 1,2,3,4,10,10-hexachloro-1,4,4a,5,8,8a- hexahydro-endo-1,2-exo-5,8- dimethanonaphthalene -- 0.25 107186 S Allyl alcohol 0.5 1.25 4 10 107051 Allyl chloride 1 3 2 6 106923 S Allyl glycidyl ether; AGE 0.2 0.93 2179591 Allyl propyl disulfide 2 12 3 18 1344281 Alumina; see Particulates not otherwise regulated Aluminum, alkyls (not otherwise classified) -- 2 Aluminum soluble salts -- 2 Aluminum metal and oxide -- Total dust -- 10 Respirable fraction(n) -- 5(n) Aluminum pyro powders -- 5 Aluminum welding fumes -- 5 300925 Aluminum distearate -- 10 7047849 Aluminum stearate -- 10 637127 Aluminum tristearate -- 10 1300738 Aminodimethylbenzene; -

Technical Assistance, Environmental Liability Clarification Or Post-Closure Modification Request Form 4400-237 (R 12/18) Page 2 Of? Section 1
State of Wisconsin Technical Assistance, Environmental Liability Department of Natural Resources PO Box 7921 , Madison WI 53707-7921 Clarification or Post-Closure Modification Request Qru,__~ Form 4400-237 (R 12/18) Page 1 of? Notice: Use this form to request a written response (on agency letterhead) from the Department of Natural Resources (DNR) regarding technical assistance, a post-closure change to a site, a specialized agreement or liability clarification for Property with known or suspected environmental contamination. A fee will be required as is authorized by s. 292.55, Wis. Stats., and NR 749, Wis. Adm. Code., unless noted in the instructions below. Personal information collected will be used for administrative purposes and may be provided to requesters to the extent required by Wisconsin's Open Records law [ss. 19.31- 19.39, Wis. Stats.]. Definitions "Property" refers to the subject Property that is perceived to have been or has been impacted by the discharge of hazardous substances. "Liability Clarification" refers to a written determination by the Department provided in response to a request made on this form. The response clarifies whether a person is or may become liable for the environmenta l contamination of a Property, as provided ins. 292.55, Wis. Slats. "Technical Assistance" refers to the Department's assistance or comments on the planning and implementation of an environmental investigation or environmental cleanup on a Property in response to a request made on thi s form as provided in s. 292.55, Wis. Slats. "Post-closure modification" refers to changes to Property boundaries and/or continuing obligations for Properties or sites that received closure letters for which continuing obligations have been applied or where contamination remains.