UNIVERSITY of CALIFORNIA Los Angeles Theoretical Study Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Facile Large Scale Solution Synthesis of Nanostructured Iron, Nickel and Cobalt Telluride and Possible Applications Sungbum Hong Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2017 Facile large scale solution synthesis of nanostructured iron, nickel and cobalt telluride and possible applications Sungbum Hong Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Chemical Engineering Commons, Materials Science and Engineering Commons, Mechanics of Materials Commons, and the Nanoscience and Nanotechnology Commons Recommended Citation Hong, Sungbum, "Facile large scale solution synthesis of nanostructured iron, nickel and cobalt telluride and possible applications" (2017). Graduate Theses and Dissertations. 15321. https://lib.dr.iastate.edu/etd/15321 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Facile large scale solution synthesis of nanostructured iron, nickel and cobalt telluride and possible applications by Sungbum Hong A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Chemical Engineering Program of Study Committee: Yue Wu, Major Professor Zengyi Shao Xinwei Wang The student author and the program of study committee are solely responsible for the content of this thesis. The Graduate College will ensure this thesis is globally accessible and will not permit alterations after a degree is conferred. Iowa State University Ames, Iowa 2017 Copyright © Sungbum Hong, 2017. All rights reserved. ii DEDICATION In memory of my Father Hong, Neung-pyo and Mother Kwon, Kyung-hee iii TABLE OF CONTENTS Page LIST OF FIGURES .................................................................................................. -

Mossbauer Spectroscopy of the Ag-Au Chalcogenides Petzite, Fischesserite
Canadian Mineralogist Vol. 30, pp.327-333(1992) MoSSBAUERsPEcrRoscoPY oF THEAs-Au GHALGoGENIDES PETZITE,FISCHESSERITE AND UYTENBOGAARDTITE FRIEDRICH E. WAGNER Physik-DepartmentEIS, TechnischeUniversittit Milnchen, D-8046 Garching, Germany JERZY A. SAWICKI AECL Research,Cholk RiverLaboratories, Chalk River, Ontario KOJ IJO JOSEPHFRIEDL Physik-DepartmentEt|, TechnischeUniversitdt Milnchen, D-8M6 Garching,Germany JOSEPHA. MANDARINO Departmentof Mineralogy'Royal OntarioMuseum, Toronto, OntarioM5S 2C6 DONALD C. HARRIS ologicalSurvey of Canada,601 Booth Street,Ottawa, Ontario KIA OE8 ABSTRACT 1974u M<lssbauerspectra of the77.3 keV l rays o1 weremeasured aL4.2K for natural and synthetic-Ag3AuTe2 (petzite),synrh;ic Ag3AuSe2(fischesserite) and syntheticAg3AuS2 (uytenbogaardtite). All compoundsstudied exhibit iarge giadients in eleitric fi6ld at the gold nuclei, notably the laigest so far found in any gold mineral' The_isomer shiits ind electricquadrupole interactions, and in particular the similarity of theseparameters for Ag3AuS2with those of Au2S, suggestthat the gold in the Ag3AuX2 compoundsshould be consideredas monovalent. Keywords:refractory Au minerals, invisible Au, structurally bound Au, Ag-Au chalcogenides,petzite, fischesserite, uy-tenbogaardtite,l97Au Mcissbauerspectroscopy, isomer shift, electric quadrupole interaction, Hollinger mine, Timmins, Hemlo deposit, Ontario. SOMMAIRE leTAu Nous avons 6tudi6les spectresde Mrissbauerdes rayons ,y (77.3 keV) de I'isotope g6n€r€sd 4,2 K et mesur6s sur des 6chantillonsnaturels er synthetiquesde Ag3AuTe2Oetzite), et des 6chantillonssynthdtiques de AgsAuSe2 (fischesserite)et Ag3AuS2(uytenbogaardiite). Touiies compos6sfont preuve d'un gradient intensedans le champ ilectrique auiour diinuclEus des atomesd'or, et en fait le plus intensequi ait 6t6 d6couvertdans une espdceaurifbre. D'aprbs les d€placementsisombres et les interactions6lectriques quadrupolaires, et en particulier la similarit6 de ces parambtresdans le Ag3AuS2avec ceux de Au2S, I'or dans ces composdsAgAuX2 serait monovalent. -

Revised Version the Crystal Structure of Uytenbogaardtite, Ag3aus2, And
Title The crystal structure of uytenbogaardtite, Ag3AuS2, and its relationships with gold and silver sulfides-selenides Authors Bindi, L; Stanley, Christopher; Seryotkin, YV; Bakakin, VR; Pal'yanova, GA; Kokh, KA Date Submitted 2017-03-30 1 1 1237R – revised version 2 The crystal structure of uytenbogaardtite, Ag3AuS2, and its relationships 3 with gold and silver sulfides-selenides 4 1, 2 3,4 5 5 LUCA BINDI *, CHRISTOPHER J. STANLEY , YURII V. SERYOTKIN , VLADIMIR V. BAKAKIN , 3,4 3,4 6 GALINA A. PAL’YANOVA , KONSTANTIN A. KOKH 7 8 1Dipartimento di Scienze della Terra, Università di Firenze, Via G. La Pira 4, I-50121Firenze, Italy 9 2Natural History Museum, Cromwell Road, London SW7 5BD, United Kingdom 3 10 Sobolev Institute of Geology and Mineralogy of the Siberian Branch of the Russian Academy of Sciences, pr. 11 Akademika Koptyuga, 3, Novosibirsk 630090, Russia 4 12 Novosibirsk State University, Pirogova str., 2, Novosibirsk 630090, Russia 5 13 Institute of Inorganic Chemistry, Siberian Branch of the RAS, prosp. Lavrentieva 3, 630090 Novosibirsk, Russia 14 15 * e-mail address: [email protected] 16 17 Abstract 18 The crystal structure of the mineral uytenbogaardtite, a rare silver-gold sulfide, was solved 19 using intensity data collected on a crystal from the type locality, the Comstock lode, Storey 20 County, Nevada (U.S.A.). The study revealed that the structure is trigonal, space group R 3 c, 3 21 with cell parameters: a = 13.6952(5), c = 17.0912(8) Å, and V = 2776.1(2) Å . The refinement 22 of an anisotropic model led to an R index of 0.0140 for 1099 independent reflections. -

Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions
minerals Article Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions Sergey Vorobyev 1, Maxim Likhatski 1, Alexander Romanchenko 1, Nikolai Maksimov 1, Sergey Zharkov 2,3, Alexander Krylov 2 and Yuri Mikhlin 1,* 1 Institute of Chemistry and Chemical Technology of the Siberian Branch of the Russian Academy of Sciences, Krasnoyarsk 660036, Russia; [email protected] (S.V.); [email protected] (M.L.); [email protected] (A.R.); [email protected] (N.M.) 2 Kirensky Institute of Physics of the Siberian Branch of the Russian Academy of Sciences, Krasnoyarsk 660036, Russia; [email protected] (S.Z.); [email protected] (A.K.) 3 Electron Microscopy Laboratory, Siberian Federal University, Krasnoyarsk 660041, Russia * Correspondence: [email protected]; Tel.: +7-391-205-1928 Received: 11 October 2018; Accepted: 29 October 2018; Published: 31 October 2018 Abstract: The reactions of aqueous gold complexes with H2Se and H2S are important for transportation and deposition of gold in nature and for synthesis of AuSe-based nanomaterials but are scantily understood. Here, we explored species formed at different proportions of HAuCl4, H2Se and H2S at room temperature using in situ UV-vis spectroscopy, dynamic light scattering (DLS), zeta-potential measurement and ex situ Transmission electron microscopy (TEM), electron diffraction, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy. Metal gold colloids arose at the molar ratios H2Se(H2S)/HAuCl4 less than 2. At higher ratios, pre-nucleation “dense liquid” species having the hydrodynamic diameter of 20–40 nm, zeta potential −40 mV to −50 mV, and the indirect band gap less than 1 eV derived from the UV-vis spectra grow into submicrometer droplets over several hours, followed by fractional nucleation in the interior and + coagulation of disordered gold chalcogenide. -

(Silver)-Telluride-(Selenide) Mineral Deposits
Geological and Atmospheric Sciences Publications Geological and Atmospheric Sciences 12-2009 Understanding gold-(silver)-telluride-(selenide) mineral deposits Nigel J. Cook University of Adelaide Cristiana L. Ciobanu University of Adelaide Paul G. Spry Iowa State University, [email protected] Panagiotis Voudouris University of Athens participants of IGCP-486 Follow this and additional works at: https://lib.dr.iastate.edu/ge_at_pubs Part of the Geochemistry Commons, Geology Commons, and the Mineral Physics Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ ge_at_pubs/354. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Geological and Atmospheric Sciences at Iowa State University Digital Repository. It has been accepted for inclusion in Geological and Atmospheric Sciences Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Understanding gold-(silver)-telluride-(selenide) mineral deposits Abstract Gold-(silver)-telluride (selenide) ores occur as epithermal orogenic and intrusion related deposits. Although Te and Se are chalcophile elements and share geochemical affinity withu, A formation of selenides and other elements Ag-Au require acidic or reducing environments. The thermodynamic stability conditions for Au and Agtellurides and native tellurium indicate an epithermal environment. Analysis of mineral paragenensis, textures and compositional variation in tellurides/selenides suggest petrogenetic processes involving interaction with fluids leading ot Au scavenging and entrapment in tellurides, changes in chemistry/rates of fluid infiltration and attaining equilibrium in a given assemblage. -

Hydrothermal Studies on Mineral Replacement Reaction in the Gold-Silver-Tellurium and Copper-Iron-Sulphur Systems
HYDROTHERMAL STUDIES ON MINERAL REPLACEMENT REACTIONS IN THE GOLD-SILVER- TELLURIUM AND COPPER-IRON-SULFR SYSTEMS JING ZHAO !" !#$% &'"( "%" !) " *+$# , ) " )#" TABLE OF CONTENTS ABSTRACT .............................................................................................................................. v DECLARATION .................................................................................................................. vii ACKNOWLEDGEMENTS .................................................................................................. ix CHAPTER 1INTRODUCTION .......................................................................................... 1 1.1 Mineral replacement reaction and coupled dissolution reprecipitation mechanism ......... 4 1.1.1 Pseudomorphism .................................................................................................................. 8 1.1.2 Porosity and fine cracks .................................................................................................... 10 1.2 Mineral replacement reactions among gold-(silver)-tellurides ....................................... 13 1.3 Mineral replacement reactions among copper iron sulfides ........................................... 17 1.4 Research objects .............................................................................................................. 23 1.5 References ....................................................................................................................... 24 CHAPTER -

Synthesis of Manganese(II) Containing Metal Chalcogen Cluster Complexes from Metal Trimethylsilylthiolate Precursors
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 2-7-2017 12:00 AM Synthesis of Manganese(II) Containing Metal Chalcogen Cluster Complexes from Metal Trimethylsilylthiolate Precursors Kyle NW Rozic The University of Western Ontario Supervisor John F. Corrigan The University of Western Ontario Graduate Program in Chemistry A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Kyle NW Rozic 2017 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Inorganic Chemistry Commons Recommended Citation Rozic, Kyle NW, "Synthesis of Manganese(II) Containing Metal Chalcogen Cluster Complexes from Metal Trimethylsilylthiolate Precursors" (2017). Electronic Thesis and Dissertation Repository. 4468. https://ir.lib.uwo.ca/etd/4468 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract The manganese(II)-palladium(II)-sulfide complex [MnCl2(µ3-S)2Pd2(dppp)2] 2 has been isolated from the reaction of [(dppp)PdCl2] with [Li(N,N’-tmeda)]2[Mn(SSiMe3)4] 1 in a 2:1 ratio under mild conditions. The trimethylsilyl thiolate complex [(dppp)Pd(SSiMe3)2] 3 has been synthesized from the reaction of [(dppp)PdCl2] with Li[SSiMe3] as well as the reaction of [(dppp)Pd(OAc)2] with Li[SSiMe3] under mild conditions. The newly synthesized complex [(dppp)Pd(SSiMe3)2] 3 was used in reaction with the manganese(II) salt [(CH3CN)2Mn(OTf)2] to form the manganese(II)-palladium(II)-sulfide complex [Mn(OTf)(thf)2(µ-S)2Pd2(dppp)2]OTf 4. -

Silver and Gold Coating
Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org Gold & Silver Coatings By A . T . Kakhia 1 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org 2 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org Part One General Knowledge 3 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org 4 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org Aqua Regia ( Royal Acid ) Freshly prepared aqua regia is colorless, Freshly prepared aqua but it turns orange within seconds. Here, regia to remove metal fresh aqua regia has been added to these salt deposits. NMR tubes to remove all traces of organic material. Contents 1 Introduction 2 Applications 3 Chemistry 3.1 Dissolving gold 3.2 Dissolving platinum 3.3 Reaction with tin 3.4 Decomposition of aqua regia 4 History 1 - Introduction Aqua regia ( Latin and Ancient Italian , lit. "royal water"), aqua regis ( Latin, lit. "king's water") , or nitro – hydro chloric acid is a highly corrosive mixture of acids, a fuming yellow or red solution. The mixture is formed by freshly mixing concentrated nitric acid and hydro chloric acid , optimally in a volume ratio of 1:3. It was named 5 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org so because it can dissolve the so - called royal or noble metals, gold and platinum. However, titanium, iridium, ruthenium, tantalum, osmium, rhodium and a few other metals are capable of with standing its corrosive properties. IUPAC name Nitric acid hydro chloride Other names aqua regia , Nitro hydrochloric acid Molecular formula HNO3 + 3 H Cl Red , yellow or gold Appearance fuming liquid 3 Density 1.01–1.21 g / cm Melting point − 42 °C Boiling point 108 °C Solubility in water miscible in water Vapor pressure 21 mbar 2 – Applications Aqua regia is primarily used to produce chloro auric acid, the electrolyte in the Wohl will process. -
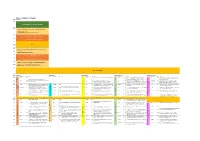
SGA Draft Schedule
Day 1 - Tuesday 27th August Room Bute Hall Time 09:00 Opening Ceremony, Welcome Addresses 09:30 Plenary 1: Geoscience vision for future exploration and resource extraction. Cam McCuaig, Principal Geoscientist, BHP 10:00 Presentation of SGA-NEWMONT Gold Medal 10:15 Presentation of SGA Young Scientist Award 10:30 10:45 Break 11:00 Plenary 2: Responsibility in Mining: How to make difficult 11:15 decisions. Sarah Gordon, Satarla Ltd, UK 11:30 Presentation of SGA-KGHM Krol Medal 11:45 Presentation Mineralium Deposita Best Paper Award 12:00 Plenary 3: Hydrothermal Origins of Brucite, Banded Iron 12:15 Formations, Transition Metal Sulfides and Life Mike Russell, Jet Propulsion Laboratory, CalTec. 12:30 Lunch and Posters 1 Room Bute Hall East Quad LT Kelvin Gallery James Watt S. J15 Stevenson LT (JWS) Time Theme S Presenter* P# Title Theme S Presenter P# Title Theme S Presenter P# Title Theme S Presenter P# Title Theme S Presenter P# Title 14:00 Kay 344 Bauer 180 The structural setting of the Barsele Au deposit, Sweden S Yajioui 13 New occurrence of Ag-Hg-Cu mineralization in the Bath 509 The critical role of architecture in constraining fluid flow Tassafte area, NE edge of the Saghro inlier, Ediacaran- and sustain chemical gradients in Late Archean Gold KEYNOTE: Andean Arc and Backarc Magmatic and Cambrian transition (Eastern Anti-Atlas, Morocco) Systems, eastern Yilgarn Tectonic Processes and the Formation of Giant Cu, Au and 14:15 Kay 344 S Perret 11 The structural control on the gold mineralization at the Li 32 The Xiasai vein-type Ag–Pb–Zn -

About the Editor
About the Editor Fritz Scholz is a professor at the University of Greifswald in Germany. Following studies in chemistry at Humboldt University, Berlin, he obtained a Dr. rer. nat. and a Dr. sc. nat. (habilitation) from that same university. In 1987 and 1989, he worked with Alan Bond in Australia. His main interest is in electrochemistry and electroanalysis. He has published more than 250 scientific papers, and he is editor and co-author of the book “Electroanalytical Methods” (Springer 2002 and 2005; Russian Edition, BINOM 2006), co-author of the book “Electrochemistry of Immobilized Particles and Droplets” (Springer 2005), co-editor of the “Electrochemical Dictionary” (Springer 2008), and co-editor of volumes 7a and 7b of the “Encyclopedia of Electrochemistry” (Wiley-VCH 2006). In 1997, he founded the Journal of Solid State Electrochemistry (Springer) and has served as Editor-in-Chief since then. He served as editor of the series “Monographs in Electrochemistry” (Springer), in which modern topics of electrochemistry are presented. Scholz introduced the technique “Voltammetry of Immobilized Microparticles” for studying the electrochemistry of solid compounds and materi- als, and he introduced the concept of threephase electrodes to determine the Gibbs energies of ion transfer between immiscible liquids. M. Bouroushian, Electrochemistry of Metal Chalcogenides, Monographs 351 in Electrochemistry, DOI 10.1007/978-3-642-03967-6, C Springer-Verlag Berlin Heidelberg 2010 About the Author Mirtat Bouroushian received a PhD for the electrodeposition and characterization of binary and ternary selenides and tellurides, from the National Technical University of Athens (NTUA; Greece, 1998). He is currently Assistant Professor of Solid State Chemistry in the Chemical Engineering School of NTUA. -

Comparison of Gold Bonding with Mercury Bonding*
CROATICA CHEMICA ACTA CCACAA, ISSN-0011-1643, ISSN-1334-417X Croat. Chem. Acta 82 (1) (2009) 233-243. CCA-3312 Original Scientific Paper Comparison of Gold Bonding with Mercury Bonding* Elfi Kraka,a Michael Filatov,b and Dieter Cremera,** aDepartment of Chemistry, University of the Pacific, 3601 Pacific Avenue, Stockton, CA 95211, USA bDepartment of Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, Netherlands RECEIVED MAY 2, 2008; REVISED AUGUST 8, 2008; ACCEPTED SEPTEMBER 9, 2008 Abstract. Nine AuX molecules (X = H, O, S, Se, Te, F, Cl, Br, I), their isoelectronic HgX+ analogues, and the corresponding neutral HgX diatomics have been investigated using NESC (Normalized Elimination of the Small Component) and B3LYP theory to determine relativistic effects for bond dissociation energies (BDEs), bond lengths, dipole moments, and charge distributions. Relativistic effects are substantially larger for AuX than HgX molecules. AuX bonding has been contrasted with HgX bonding considering the effects of relativity, charge transfer and ionic bonding, 3-electron versus 2-electron bonding, residual π- bonding, lone pair repulsion, and the d-block effect. The interplay of the various electronic effects leads to strongly differing trends in calculated BDEs, which can be rationalized with a simple MO model based on electronegativity differences, atomic orbital energies, and their change due to scalar relativity. A relativis- tic increase or decrease in the BDE is directly related to relativistic -

University of Groningen Comparison of Gold Bonding With
University of Groningen Comparison of Gold Bonding with Mercury Bonding Kraka, Elfi; Filatov, Michael; Cremer, Dieter Published in: Croatica Chemica Acta IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2009 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Kraka, E., Filatov, M., & Cremer, D. (2009). Comparison of Gold Bonding with Mercury Bonding. Croatica Chemica Acta, 82(1), 233-243. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 12-11-2019 CROATICA CHEMICA ACTA CCACAA, ISSN-0011-1643, ISSN-1334-417X Croat. Chem. Acta 82 (1) (2009) 233-243. CCA-3312 Original Scientific Paper Comparison of Gold Bonding with Mercury Bonding* Elfi Kraka,a Michael Filatov,b and Dieter Cremera,** aDepartment of Chemistry, University of the Pacific, 3601 Pacific Avenue, Stockton, CA 95211, USA bDepartment of Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, Netherlands RECEIVED MAY 2, 2008; REVISED AUGUST 8, 2008; ACCEPTED SEPTEMBER 9, 2008 Abstract.