Supplemental Information Metabolic Roles of Uncultivated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

New Opportunities Revealed by Biotechnological Explorations of Extremophiles - Mircea Podar and Anna-Louise Reysenbach
BIOTECHNOLOGY – Vol .III – New Opportunities Revealed by Biotechnological Explorations of Extremophiles - Mircea Podar and Anna-Louise Reysenbach NEW OPPORTUNITIES REVEALED BY BIOTECHNOLOGICAL EXPLORATIONS OF EXTREMOPHILES Mircea Podar and Anna-Louise Reysenbach Department of Biology, Portland State University, Portland, OR 97201, USA. Keywords: extremophiles, genomics, biotechnology, enzymes, metagenomics. Contents 1. Introduction 2. Extremophiles and Biomolecules 3. Extremophile Genomics Exposing the Biotechnological Potential 4. Tapping into the Hidden Biotechnological Potential through Metagenomics 5. Unexplored Frontiers and Future Prospects Acknowledgements Glossary Bibliography Biographical Sketches Summary Over the past few decades the extremes at which life thrives has continued to challenge our understanding of biochemistry, biology and evolution. As more new extremophiles are brought into laboratory culture, they have provided a multitude of new potential applications for biotechnology. Furthermore, more recently, innovative culturing approaches, environmental genome sequencing and whole genome sequencing have provided new opportunities for biotechnological exploration of extremophiles. 1. Introduction Organisms that live at the extremes of pH (>pH 8.5,< pH 5.0), temperature (>45°C, <15°C), pressure (>500 atm), salinity (>1.0M NaCl) and in high concentrations of recalcitrant substances or heavy metals (extremophiles) represent one of the last frontiers for biotechnological and industrial discovery. As we learn more about the -
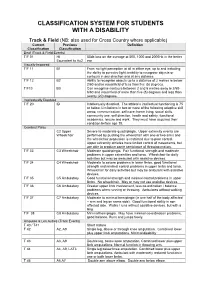
Disability Classification System
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

Eelgrass Sediment Microbiome As a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan
Microbes Environ. Vol. 34, No. 1, 13-22, 2019 https://www.jstage.jst.go.jp/browse/jsme2 doi:10.1264/jsme2.ME18103 Eelgrass Sediment Microbiome as a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan TATSUNORI NAKAGAWA1*, YUKI TSUCHIYA1, SHINGO UEDA1, MANABU FUKUI2, and REIJI TAKAHASHI1 1College of Bioresource Sciences, Nihon University, 1866 Kameino, Fujisawa, 252–0880, Japan; and 2Institute of Low Temperature Science, Hokkaido University, Kita-19, Nishi-8, Kita-ku, Sapporo, 060–0819, Japan (Received July 16, 2018—Accepted October 22, 2018—Published online December 1, 2018) Nitrous oxide (N2O) is a powerful greenhouse gas; however, limited information is currently available on the microbiomes involved in its sink and source in seagrass meadow sediments. Using laboratory incubations, a quantitative PCR (qPCR) analysis of N2O reductase (nosZ) and ammonia monooxygenase subunit A (amoA) genes, and a metagenome analysis based on the nosZ gene, we investigated the abundance of N2O-reducing microorganisms and ammonia-oxidizing prokaryotes as well as the community compositions of N2O-reducing microorganisms in in situ and cultivated sediments in the non-eelgrass and eelgrass zones of Lake Akkeshi, Japan. Laboratory incubations showed that N2O was reduced by eelgrass sediments and emitted by non-eelgrass sediments. qPCR analyses revealed that the abundance of nosZ gene clade II in both sediments before and after the incubation as higher in the eelgrass zone than in the non-eelgrass zone. In contrast, the abundance of ammonia-oxidizing archaeal amoA genes increased after incubations in the non-eelgrass zone only. Metagenome analyses of nosZ genes revealed that the lineages Dechloromonas-Magnetospirillum-Thiocapsa and Bacteroidetes (Flavobacteriia) within nosZ gene clade II were the main populations in the N2O-reducing microbiome in the in situ sediments of eelgrass zones. -

Localizing Transcripts to Single Cells Suggests an Important Role of Uncultured Deltaproteobacteria in the Termite Gut Hydrogen Economy
Localizing transcripts to single cells suggests an important role of uncultured deltaproteobacteria in the termite gut hydrogen economy Adam Z. Rosenthala,1, Xinning Zhanga,1, Kaitlyn S. Luceya, Elizabeth A. Ottesena, Vikas Trivedib, Harry M. T. Choib, Niles A. Pierceb,c, and Jared R. Leadbettera,2 aRonald and Maxine Linde Center for Global Environmental Science, bDepartment of Bioengineering, and cDepartment of Computing and Mathematical Sciences, California Institute of Technology, Pasadena, CA 91125 Edited by James M. Tiedje, Michigan State University, East Lansing, MI, and approved August 13, 2013 (received for review April 29, 2013) Identifying microbes responsible for particular environmental and in situ assays to address such matters directly. We interro- functions is challenging, given that most environments contain gated a tiny, yet complex environment that accommodates robust, an uncultivated microbial diversity. Here we combined approaches stable, and species-rich microbial communities—the hindgut of a to identify bacteria expressing genes relevant to catabolite flow wood-feeding lower termite, Zootermopsis nevadensis (1). and to locate these genes within their environment, in this case Termites and their gut microbiota digest lignocellulose, the the gut of a “lower,” wood-feeding termite. First, environmental most abundant natural composite material on Earth. For some time now, it has been known that a key activity in this nutritional transcriptomics revealed that 2 of the 23 formate dehydrogenase + (FDH) genes known in the system accounted for slightly more than mutualism involves the bacterial conversion of H2 CO2, gen- one-half of environmental transcripts. FDH is an essential enzyme erated during wood polysaccharide fermentation, into acetate in a process called CO -reductive acetogenesis (2, 3). -

The Sunlit Microoxic Niche of the Archaeal Eukaryotic Ancestor Comes 2 to Light
bioRxiv preprint doi: https://doi.org/10.1101/385732; this version posted August 20, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 The sunlit microoxic niche of the archaeal eukaryotic ancestor comes 2 to light 3 4 Paul-Adrian Bulzu1#, Adrian-Ştefan Andrei2#, Michaela M. Salcher3, Maliheh Mehrshad2, 5 Keiichi Inoue5, Hideki Kandori6, Oded Beja4, Rohit Ghai2*, Horia L. Banciu1,7 6 1Department of Molecular Biology and Biotechnology, Faculty of Biology and Geology, Babeş-Bolyai 7 University, Cluj-Napoca, Romania 8 2Institute of Hydrobiology, Department of Aquatic Microbial Ecology, Biology Centre of the Academy 9 of Sciences of the Czech Republic, České Budějovice, Czech Republic. 10 3Limnological Station, Institute of Plant and Microbial Biology, University of Zurich, Seestrasse 187, 11 CH-8802 Kilchberg, Switzerland. 12 4Faculty of Biology, Technion Israel Institute of Technology, Haifa, Israel. 13 5The Institute for Solid State Physics, The University of Tokyo, Kashiwa, Japan 14 6Department of Life Science and Applied Chemistry, Nagoya Institute of Technology, Nagoya, Japan. 15 7Molecular Biology Center, Institute for Interdisciplinary Research in Bio-Nano-Sciences, Babeş- 16 Bolyai University, Cluj-Napoca, Romania 17 18 19 20 21 # These authors contributed equally to this work 22 *Corresponding author: Rohit Ghai 23 Institute of Hydrobiology, Department of Aquatic Microbial Ecology, Biology Centre of the Academy 24 of Sciences of the Czech Republic, Na Sádkách 7, 370 05, České Budějovice, Czech Republic. -

Instruction Manual
EN7528563-00 07 - 2017 LGN power DYNACIAT Instruction manual EN CONTENTS PAGE 1 INTRODUCTION 2 15 INSTALLATION ANTIFREEZE PROTECTION 13 2 SHIPMENT OF THE UNIT 3 16 ELECTRICAL CONNECTIONS 13 3 RECEIPT OF GOODS 3 16.1 Power connection 13 3.1 Checking the equipment 3 16.2 Connection to the air-cooled condenser. 14 3.2 Identifying the equipment 3 16.3 Customer connection for remote 4 SAFETY INSTRUCTIONS 4 control functions. 15 5 MACHINE COMPLIANCE 4 17 CONTROL AND SAFETY DEVICES 16 17.1 Electronic control and display module 16 6 WARRANTY 4 17.2 Main functions 16 7 UNIT LOCATION 4 17.3 Safety device management 16 8 HANDLING AND POSITIONING 5 17.4 Phase controller kit 17 17.5 Location of the safety sensors and devices 18 9 LOCATION 6 17.6 Adjusting the control and safety devices 19 9.1 Location of the unit 6 18 COMMISSIONING 19 10 OPERATING LIMITS 7 18.1 Commissioning 19 10.1 Operating range 7 18.2 Essential points that must be checked 20 10.2 Limits 7 19 TECHNICAL CHARACTERISTICS 21 10.3 Evaporator limits 7 10.4 Minimum/maximum water flow rates 7 20 ELECTRICAL SPECIFICATIONS 22 11 LOCATION OF THE MAIN COMPONENTS 8 21. SERVICING AND MAINTENANCE 23 12 MAIN COMPONENTS OF THE 21.1 Operating readings 23 REFRIGERATING CIRCUIT 8 21.2 Unit maintenance and servicing 23 13 HYDRAULIC CONNECTIONS 9 22 ECODESIGN 25 13.1 Diameters of the hydraulic connections 9 23 PERMANENT SHUTDOWN 25 13.2 Connections 10 24 TROUBLESHOOTING OPERATING 13.3 FLANGE/VICTAULIC adapter kit (OPTION) 10 PROBLEMS 26 14 REFRIGERANT CONNECTIONS 11 25 SYSTEM SCHEMATICS 27 14.1 General information 11 25.1 ooling installation with drycooler 27 14.2 Accessories 11 14.3 Routing and sizing 11 14.4 Installation 12 ORIGINAL TEXT: FRENCH VERSION EN - 1 1 INTRODUCTION DYNACIATpower LGN series water chillers are designed to meet the air conditioning requirements of residential and office buildings as well as the requirements of manufacturing processes. -

Geomicrobiological Processes in Extreme Environments: a Review
202 Articles by Hailiang Dong1, 2 and Bingsong Yu1,3 Geomicrobiological processes in extreme environments: A review 1 Geomicrobiology Laboratory, China University of Geosciences, Beijing, 100083, China. 2 Department of Geology, Miami University, Oxford, OH, 45056, USA. Email: [email protected] 3 School of Earth Sciences, China University of Geosciences, Beijing, 100083, China. The last decade has seen an extraordinary growth of and Mancinelli, 2001). These unique conditions have selected Geomicrobiology. Microorganisms have been studied in unique microorganisms and novel metabolic functions. Readers are directed to recent review papers (Kieft and Phelps, 1997; Pedersen, numerous extreme environments on Earth, ranging from 1997; Krumholz, 2000; Pedersen, 2000; Rothschild and crystalline rocks from the deep subsurface, ancient Mancinelli, 2001; Amend and Teske, 2005; Fredrickson and Balk- sedimentary rocks and hypersaline lakes, to dry deserts will, 2006). A recent study suggests the importance of pressure in the origination of life and biomolecules (Sharma et al., 2002). In and deep-ocean hydrothermal vent systems. In light of this short review and in light of some most recent developments, this recent progress, we review several currently active we focus on two specific aspects: novel metabolic functions and research frontiers: deep continental subsurface micro- energy sources. biology, microbial ecology in saline lakes, microbial Some metabolic functions of continental subsurface formation of dolomite, geomicrobiology in dry deserts, microorganisms fossil DNA and its use in recovery of paleoenviron- Because of the unique geochemical, hydrological, and geological mental conditions, and geomicrobiology of oceans. conditions of the deep subsurface, microorganisms from these envi- Throughout this article we emphasize geomicrobiological ronments are different from surface organisms in their metabolic processes in these extreme environments. -

Sporulation Evolution and Specialization in Bacillus
bioRxiv preprint doi: https://doi.org/10.1101/473793; this version posted March 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Research article From root to tips: sporulation evolution and specialization in Bacillus subtilis and the intestinal pathogen Clostridioides difficile Paula Ramos-Silva1*, Mónica Serrano2, Adriano O. Henriques2 1Instituto Gulbenkian de Ciência, Oeiras, Portugal 2Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Oeiras, Portugal *Corresponding author: Present address: Naturalis Biodiversity Center, Marine Biodiversity, Leiden, The Netherlands Phone: 0031 717519283 Email: [email protected] (Paula Ramos-Silva) Running title: Sporulation from root to tips Keywords: sporulation, bacterial genome evolution, horizontal gene transfer, taxon- specific genes, Bacillus subtilis, Clostridioides difficile 1 bioRxiv preprint doi: https://doi.org/10.1101/473793; this version posted March 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Bacteria of the Firmicutes phylum are able to enter a developmental pathway that culminates with the formation of a highly resistant, dormant spore. Spores allow environmental persistence, dissemination and for pathogens, are infection vehicles. In both the model Bacillus subtilis, an aerobic species, and in the intestinal pathogen Clostridioides difficile, an obligate anaerobe, sporulation mobilizes hundreds of genes. -

Download/Issues/Mining/Reference Guide to Treatment Technologi Es for MIW.Pdf
Removal of Soluble Selenium in the Presence of Nitrate from Coal Mining-Influenced Water by Frank Nkansah - Boadu BSc., Kwame Nkrumah University of Science and Technology, 2003 MASc., The University of British Columbia, 2013 A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Chemical and Biological Engineering) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) December 2019 © Frank Nkansah - Boadu, 2019 The following individuals certify that they have read, and recommend to the Faculty of Graduate and Postdoctoral Studies for acceptance, the dissertation entitled: Removal of Soluble Selenium in the Presence of Nitrate from Coal Mining-Influenced Water submitted by Frank Nkansah-Boadu in partial fulfillment of the requirements for the degree of Doctor of Philosophy In Chemical and Biological Engineering Examining Committee: Susan Baldwin, Chemical and Biological Engineering Supervisor Vikramaditya Yadav, Chemical and Biological Engineering Supervisory Committee Member Troy Vassos, Adjunct Professor, Civil Engineering Supervisory Committee Member Anthony Lau, Chemical and Biological Engineering University Examiner Scott Dunbar, Mining Engineering University Examiner ii Abstract Biological treatment to remove dissolved selenium from mining-influenced water (MIW) is inhibited by co-contaminants, especially nitrate. It was hypothesized that selenium reducing microorganisms can be obtained from native mine bacteria at sites affected by MIW due to the selection pressure from elevated selenium concentrations at those sites. Enrichment of these microorganisms and testing of their capacity to remove dissolved selenium from actual coal MIW was the objective of this dissertation. Fifteen sediments were collected from eleven different vegetated or non-vegetated seepage collection ponds and one non-impacted natural wetland. -

Mannoside Recognition and Degradation by Bacteria Simon Ladeveze, Elisabeth Laville, Jordane Despres, Pascale Mosoni, Gabrielle Veronese
Mannoside recognition and degradation by bacteria Simon Ladeveze, Elisabeth Laville, Jordane Despres, Pascale Mosoni, Gabrielle Veronese To cite this version: Simon Ladeveze, Elisabeth Laville, Jordane Despres, Pascale Mosoni, Gabrielle Veronese. Mannoside recognition and degradation by bacteria. Biological Reviews, Wiley, 2016, 10.1111/brv.12316. hal- 01602393 HAL Id: hal-01602393 https://hal.archives-ouvertes.fr/hal-01602393 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biol. Rev. (2016), pp. 000–000. 1 doi: 10.1111/brv.12316 Mannoside recognition and degradation by bacteria Simon Ladeveze` 1, Elisabeth Laville1, Jordane Despres2, Pascale Mosoni2 and Gabrielle Potocki-Veron´ ese` 1∗ 1LISBP, Universit´e de Toulouse, CNRS, INRA, INSA, 31077, Toulouse, France 2INRA, UR454 Microbiologie, F-63122, Saint-Gen`es Champanelle, France ABSTRACT Mannosides constitute a vast group of glycans widely distributed in nature. Produced by almost all organisms, these carbohydrates are involved in numerous cellular processes, such as cell structuration, protein maturation and signalling, mediation of protein–protein interactions and cell recognition. The ubiquitous presence of mannosides in the environment means they are a reliable source of carbon and energy for bacteria, which have developed complex strategies to harvest them. -

Protistology an International Journal Vol
Protistology An International Journal Vol. 10, Number 2, 2016 ___________________________________________________________________________________ CONTENTS INTERNATIONAL SCIENTIFIC FORUM «PROTIST–2016» Yuri Mazei (Vice-Chairman) Welcome Address 2 Organizing Committee 3 Organizers and Sponsors 4 Abstracts 5 Author Index 94 Forum “PROTIST-2016” June 6–10, 2016 Moscow, Russia Website: http://onlinereg.ru/protist-2016 WELCOME ADDRESS Dear colleagues! Republic) entitled “Diplonemids – new kids on the block”. The third lecture will be given by Alexey The Forum “PROTIST–2016” aims at gathering Smirnov (Saint Petersburg State University, Russia): the researchers in all protistological fields, from “Phylogeny, diversity, and evolution of Amoebozoa: molecular biology to ecology, to stimulate cross- new findings and new problems”. Then Sandra disciplinary interactions and establish long-term Baldauf (Uppsala University, Sweden) will make a international scientific cooperation. The conference plenary presentation “The search for the eukaryote will cover a wide range of fundamental and applied root, now you see it now you don’t”, and the fifth topics in Protistology, with the major focus on plenary lecture “Protist-based methods for assessing evolution and phylogeny, taxonomy, systematics and marine water quality” will be made by Alan Warren DNA barcoding, genomics and molecular biology, (Natural History Museum, United Kingdom). cell biology, organismal biology, parasitology, diversity and biogeography, ecology of soil and There will be two symposia sponsored by ISoP: aquatic protists, bioindicators and palaeoecology. “Integrative co-evolution between mitochondria and their hosts” organized by Sergio A. Muñoz- The Forum is organized jointly by the International Gómez, Claudio H. Slamovits, and Andrew J. Society of Protistologists (ISoP), International Roger, and “Protists of Marine Sediments” orga- Society for Evolutionary Protistology (ISEP), nized by Jun Gong and Virginia Edgcomb. -

Thermophilic Carboxydotrophs and Their Applications in Biotechnology Springerbriefs in Microbiology
SPRINGER BRIEFS IN MICROBIOLOGY EXTREMOPHILIC BACTERIA Sonia M. Tiquia-Arashiro Thermophilic Carboxydotrophs and their Applications in Biotechnology SpringerBriefs in Microbiology Extremophilic Bacteria Series editors Sonia M. Tiquia-Arashiro, Dearborn, MI, USA Melanie Mormile, Rolla, MO, USA More information about this series at http://www.springer.com/series/11917 Sonia M. Tiquia-Arashiro Thermophilic Carboxydotrophs and their Applications in Biotechnology 123 Sonia M. Tiquia-Arashiro Department of Natural Sciences University of Michigan Dearborn, MI USA ISSN 2191-5385 ISSN 2191-5393 (electronic) ISBN 978-3-319-11872-7 ISBN 978-3-319-11873-4 (eBook) DOI 10.1007/978-3-319-11873-4 Library of Congress Control Number: 2014951696 Springer Cham Heidelberg New York Dordrecht London © The Author(s) 2014 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifically for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. Duplication of this publication or parts thereof is permitted only under the provisions of the Copyright Law of the Publisher’s location, in its current version, and permission for use must always be obtained from Springer.