Gene Section Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Mouse Germ Line Mutations Due to Retrotransposon Insertions Liane Gagnier1, Victoria P
Gagnier et al. Mobile DNA (2019) 10:15 https://doi.org/10.1186/s13100-019-0157-4 REVIEW Open Access Mouse germ line mutations due to retrotransposon insertions Liane Gagnier1, Victoria P. Belancio2 and Dixie L. Mager1* Abstract Transposable element (TE) insertions are responsible for a significant fraction of spontaneous germ line mutations reported in inbred mouse strains. This major contribution of TEs to the mutational landscape in mouse contrasts with the situation in human, where their relative contribution as germ line insertional mutagens is much lower. In this focussed review, we provide comprehensive lists of TE-induced mouse mutations, discuss the different TE types involved in these insertional mutations and elaborate on particularly interesting cases. We also discuss differences and similarities between the mutational role of TEs in mice and humans. Keywords: Endogenous retroviruses, Long terminal repeats, Long interspersed elements, Short interspersed elements, Germ line mutation, Inbred mice, Insertional mutagenesis, Transcriptional interference Background promoter and polyadenylation motifs and often a splice The mouse and human genomes harbor similar types of donor site [10, 11]. Sequences of full-length ERVs can TEs that have been discussed in many reviews, to which encode gag, pol and sometimes env, although groups of we refer the reader for more in depth and general infor- LTR retrotransposons with little or no retroviral hom- mation [1–9]. In general, both human and mouse con- ology also exist [6–9]. While not the subject of this re- tain ancient families of DNA transposons, none view, ERV LTRs can often act as cellular enhancers or currently active, which comprise 1–3% of these genomes promoters, creating chimeric transcripts with genes, and as well as many families or groups of retrotransposons, have been implicated in other regulatory functions [11– which have caused all the TE insertional mutations in 13]. -

Transcriptional Mechanisms of Resistance to Anti-PD-1 Therapy
Author Manuscript Published OnlineFirst on February 13, 2017; DOI: 10.1158/1078-0432.CCR-17-0270 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Transcriptional mechanisms of resistance to anti-PD-1 therapy Maria L. Ascierto1, Alvin Makohon-Moore2, 11, Evan J. Lipson1, Janis M. Taube3,4, Tracee L. McMiller5, Alan E. Berger6, Jinshui Fan6, Genevieve J. Kaunitz3, Tricia R. Cottrell4, Zachary A. Kohutek7, Alexander Favorov8,10, Vladimir Makarov7,11, Nadeem Riaz7,11, Timothy A. Chan7,11, Leslie Cope8, Ralph H. Hruban4,9, Drew M. Pardoll1, Barry S. Taylor11,12,13, David B. Solit13, Christine A Iacobuzio-Donahue2,11, and Suzanne L. Topalian5 From the 1Departments of Oncology, 3Dermatology, 4Pathology, 5Surgery, 6The Lowe Family Genomics Core, 8Oncology Bioinformatics Core, and the 9 Sol Goldman Pancreatic Cancer Research Center, Johns Hopkins University School of Medicine and Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD 21287; the 10Laboratory of System Biology and Computational Genetics, Vavilov Institute of General Genetics, Russian Academy of Sciences, 119991, Moscow, Russia; and 2Pathology, 7Radiation Oncology, 11Human Oncology and Pathogenesis Program, 12Department of Epidemiology and Biostatistics, and the 13Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center, New York NY 10065. MLA, AM-M, EJL, and JMT contributed equally to this work Running title: Transcriptional mechanisms of resistance to anti-PD-1 Key Words: melanoma, cancer genetics, immunotherapy, anti-PD-1 Financial Support: This study was supported by the Melanoma Research Alliance (to SLT and CI-D), the Bloomberg~Kimmel Institute for Cancer Immunotherapy (to JMT, DMP, and SLT), the Barney Family Foundation (to SLT), Moving for Melanoma of Delaware (to SLT), the 1 Downloaded from clincancerres.aacrjournals.org on October 2, 2021. -

Type of the Paper (Article
Cancers 2020, 12, S1 of S11 Supplementary Materials: Inflammatory Proteins HMGA2 and PRTN3 as Drivers of Vulvar Squamous Cell Carcinoma Pro- gression Agnieszka Fatalska, Natalia Rusetska, Elwira Bakuła-Zalewska, Artur Kowalik, Sebastian Zięba, Agnieszka Wroblewska, Kamil Zalewski, Krzysztof Goryca, Dominik Domański and Magdalena Kowalewska Text S1: Supplementary Methods iTRAQ (Isobaric Tags for Relative and Absolute Quantitation) Cell Lysis The samples were snap-frozen in liquid nitrogen immediately after collection and stored at −70°C before pulverization (with the Microdismembrator II, B Braun Biotech In- ternational, Melsungen, Germany). Forty-two (14 control, 16 d-fVSCC and 12 progVSCC) samples were subjected to deep proteomic analysis. Total cell lysates were obtained from approximately 100 mg of pulverized patient tissue samples. The frozen pulverized tissue samples were resuspended in 300 µl of cold Lysis Buffer (1% sodium deoxycholate (NaDOC) in 25 mM HEPES (4-(2-hydroxyethyl)-1-pipera- zineethanesulfonic acid), boiled for 5 min at 100 °C, and cooled to room temperature (RT) on ice. Next, nucleic acids were degraded with 30 µL of benzonase nuclease (2.5 U/µL in 25 mM HEPES) at 4 °C for 30 min. Samples were then centrifuged twice at 12,000 × g for 10 min at 4 °C and the supernatants were stored at −80 °C. Protein concentrations were determined using the Pierce (Appleton, WI, USA) Bicinchoninic Acid (BCA) Assay Kit according to the manufacturer’s instructions, for the 96-well plate format, in duplicate, at two different sample dilutions (ThermoFisher Scientific, Waltham, MA, USA). Sample Protein Extract Digestion and iTRAQ Labeling Protein extract digestion and iTRAQ tag labeling was conducted using the 8-plex iTRAQ assay and iTRAQ Reagent and Buffer Kits (AB SCIEX, Framingham, MA, USA). -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Mapping Our Genes—Genome Projects: How Big? How Fast?
Mapping Our Genes—Genome Projects: How Big? How Fast? April 1988 NTIS order #PB88-212402 Recommended Citation: U.S. Congress, Office of Technology Assessment, Mapping Our Genes-The Genmne Projects.’ How Big, How Fast? OTA-BA-373 (Washington, DC: U.S. Government Printing Office, April 1988). Library of Congress Catalog Card Number 87-619898 For sale by the Superintendent of Documents U.S. Government Printing Office, Washington, DC 20402-9325 (order form can be found in the back of this report) Foreword For the past 2 years, scientific and technical journals in biology and medicine have extensively covered a debate about whether and how to determine the function and order of human genes on human chromosomes and when to determine the sequence of molecular building blocks that comprise DNA in those chromosomes. In 1987, these issues rose to become part of the public agenda. The debate involves science, technol- ogy, and politics. Congress is responsible for ‘(writing the rules” of what various Federal agencies do and for funding their work. This report surveys the points made so far in the debate, focusing on those that most directly influence the policy options facing the U.S. Congress, The House Committee on Energy and Commerce requested that OTA undertake the project. The House Committee on Science, Space, and Technology, the Senate Com- mittee on Labor and Human Resources, and the Senate Committee on Energy and Natu- ral Resources also asked OTA to address specific points of concern to them. Congres- sional interest focused on several issues: ● how to assess the rationales for conducting human genome projects, ● how to fund human genome projects (at what level and through which mech- anisms), ● how to coordinate the scientific and technical programs of the several Federal agencies and private interests already supporting various genome projects, and ● how to strike a balance regarding the impact of genome projects on international scientific cooperation and international economic competition in biotechnology. -

Genetic Variant in 3' Untranslated Region of the Mouse Pycard Gene
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 3 Title: 4 Genetic Variant in 3’ Untranslated Region of the Mouse Pycard Gene Regulates Inflammasome 5 Activity 6 Running Title: 7 3’UTR SNP in Pycard regulates inflammasome activity 8 Authors: 9 Brian Ritchey1*, Qimin Hai1*, Juying Han1, John Barnard2, Jonathan D. Smith1,3 10 1Department of Cardiovascular & Metabolic Sciences, Lerner Research Institute, Cleveland Clinic, 11 Cleveland, OH 44195 12 2Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 13 44195 14 3Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine of Case Western 15 Reserve University, Cleveland, OH 44195 16 *, These authors contributed equally to this study. 17 Address correspondence to Jonathan D. Smith: email [email protected]; ORCID ID 0000-0002-0415-386X; 18 mailing address: Cleveland Clinic, Box NC-10, 9500 Euclid Avenue, Cleveland, OH 44195, USA. 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Quantitative trait locus mapping for interleukin-1 release after inflammasome priming and activation 22 was performed on bone marrow-derived macrophages (BMDM) from an AKRxDBA/2 strain intercross. -

Atlas Journal
Atlas of Genetics and Cytogenetics in Oncology and Haematology Home Genes Leukemias Tumors Cancer prone Deep Insight Case Reports Portal Journals Teaching X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 NA Atlas Journal Atlas Journal versus Atlas Database: the accumulation of the issues of the Journal constitutes the body of the Database/Text-Book. TABLE OF CONTENTS Volume 13, Number 7, July 2009 Previous Issue / Next Issue Genes ABL1 (v-abl Abelson murine leukemia viral oncogene homolog 1) (9q34.1) - updated. Ali G Turhan. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 757-766. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/ABL.html BCL2L12 (BCL2-like 12 (proline-rich)) (19q13.3). Christos Kontos, Hellinida Thomadaki, Andreas Scorilas. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 767-771. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/BCL2L12ID773ch19q13.html BCR (Breakpoint cluster region) (22q11.2) - updated. Ali G Turhan. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 772-779. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/BCR.html ENAH (enabled homolog (Drosophila)) (1q42.12). Paola Nisticò, Francesca Di Modugno. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 780-785. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/ENAHID44148ch1q42.html FGFR2 (fibroblast growth factor receptor 2) (10q26.13). Masaru Katoh. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 786-799. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/FGFR2ID40570ch10q26.html MAPK6 (mitogen-activated protein kinase 6) (15q21.2). Sylvain Meloche. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 800-804. -

Disease-Related Cellular Protein Networks Differentially Affected
www.nature.com/scientificreports OPEN Disease‑related cellular protein networks diferentially afected under diferent EGFR mutations in lung adenocarcinoma Toshihide Nishimura1,8*, Haruhiko Nakamura1,2,8, Ayako Yachie3,8, Takeshi Hase3,8, Kiyonaga Fujii1,8, Hirotaka Koizumi4, Saeko Naruki4, Masayuki Takagi4, Yukiko Matsuoka3, Naoki Furuya5, Harubumi Kato6,7 & Hisashi Saji2 It is unclear how epidermal growth factor receptor EGFR major driver mutations (L858R or Ex19del) afect downstream molecular networks and pathways. This study aimed to provide information on the infuences of these mutations. The study assessed 36 protein expression profles of lung adenocarcinoma (Ex19del, nine; L858R, nine; no Ex19del/L858R, 18). Weighted gene co-expression network analysis together with analysis of variance-based screening identifed 13 co-expressed modules and their eigen proteins. Pathway enrichment analysis for the Ex19del mutation demonstrated involvement of SUMOylation, epithelial and mesenchymal transition, ERK/mitogen- activated protein kinase signalling via phosphorylation and Hippo signalling. Additionally, analysis for the L858R mutation identifed various pathways related to cancer cell survival and death. With regard to the Ex19del mutation, ROCK, RPS6KA1, ARF1, IL2RA and several ErbB pathways were upregulated, whereas AURK and GSKIP were downregulated. With regard to the L858R mutation, RB1, TSC22D3 and DOCK1 were downregulated, whereas various networks, including VEGFA, were moderately upregulated. In all mutation types, CD80/CD86 (B7), MHC, CIITA and IFGN were activated, whereas CD37 and SAFB were inhibited. Costimulatory immune-checkpoint pathways by B7/CD28 were mainly activated, whereas those by PD-1/PD-L1 were inhibited. Our fndings may help identify potential therapeutic targets and develop therapeutic strategies to improve patient outcomes. -

Strains Used in Whole Organism Plasmodium Falciparum Vaccine Trials Differ in 2 Genome Structure, Sequence, and Immunogenic Potential 3 4 Authors: Kara A
bioRxiv preprint doi: https://doi.org/10.1101/684175; this version posted June 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Strains used in whole organism Plasmodium falciparum vaccine trials differ in 2 genome structure, sequence, and immunogenic potential 3 4 Authors: Kara A. Moser1‡, Elliott F. Drábek1, Ankit Dwivedi1, Jonathan Crabtree1, Emily 5 M. Stucke2, Antoine Dara2, Zalak Shah2, Matthew Adams2, Tao Li3, Priscila T. 6 Rodrigues4, Sergey Koren5, Adam M. Phillippy5, Amed Ouattara2, Kirsten E. Lyke2, Lisa 7 Sadzewicz1, Luke J. Tallon1, Michele D. Spring6, Krisada Jongsakul6, Chanthap Lon6, 8 David L. Saunders6¶, Marcelo U. Ferreira4, Myaing M. Nyunt2§, Miriam K. Laufer2, Mark 9 A. Travassos2, Robert W. Sauerwein7, Shannon Takala-Harrison2, Claire M. Fraser1, B. 10 Kim Lee Sim3, Stephen L. Hoffman3, Christopher V. Plowe2§, Joana C. Silva1,8 11 12 Corresponding author: Joana C. Silva, [email protected] 13 Affiliations: 14 1 Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, 15 MD, 21201 16 2 Center for Vaccine Development and Global Health, University of Maryland School of 17 Medicine, Baltimore, MD, 21201 18 3 Sanaria, Inc. Rockville, Maryland, 20850 19 4 Department of Parasitology, Institute of Biomedical Sciences, University of São Paulo, 20 São Paulo, Brazil 21 5 Genome Informatics Section, Computational and Statistical Genomics Branch, 22 National Human -
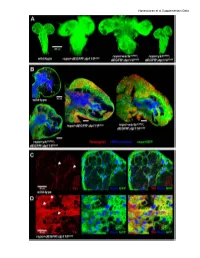
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

De Novo Profiling of Long Non-Coding Rnas Involved
International Journal of Molecular Sciences Article De Novo Profiling of Long Non-Coding RNAs Involved in MC-LR-Induced Liver Injury in Whitefish: Discovery and Perspectives Maciej Florczyk * , Paweł Brzuzan and Maciej Wo´zny Department of Environmental Biotechnology, Faculty of Geoengineering, University of Warmia and Mazury in Olsztyn, ul. Słoneczna 45G, 10-709 Olsztyn, Poland; [email protected] (P.B.); [email protected] (M.W.) * Correspondence: maciej.fl[email protected] Abstract: Microcystin-LR (MC-LR) is a potent hepatotoxin for which a substantial gap in knowledge persists regarding the underlying molecular mechanisms of liver toxicity and injury. Although long non-coding RNAs (lncRNAs) have been extensively studied in model organisms, our knowledge concerning the role of lncRNAs in liver injury is limited. Given that lncRNAs show low levels of sequence conservation, their role becomes even more unclear in non-model organisms without an annotated genome, like whitefish (Coregonus lavaretus). The objective of this study was to discover and profile aberrantly expressed polyadenylated lncRNAs that are involved in MC-LR-induced liver injury in whitefish. Using RNA sequencing (RNA-Seq) data, we de novo assembled a high-quality whitefish liver transcriptome. This enabled us to find 94 differentially expressed (DE) putative evolutionary conserved lncRNAs, such as MALAT1, HOTTIP, HOTAIR or HULC, and 4429 DE putative novel whitefish lncRNAs, which differed from annotated protein-coding transcripts (PCTs) in terms of minimum free energy, guanine-cytosine (GC) base-pair content and length. Additionally, we identified DE non-coding transcripts that might be 30 autonomous untranslated regions (30UTRs) Citation: Florczyk, M.; Brzuzan, P.; Wo´zny, M.