Polar Intermetallic Compounds of the Silicon and Arsenic Family Elements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Disability Classification System
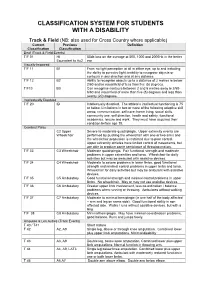
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

Identification and Quantification of Arsenic Species in Gold Mine Wastes Using Synchrotron-Based X-Ray Techniques
Identification and Quantification of Arsenic Species in Gold Mine Wastes Using Synchrotron-Based X-ray Techniques Andrea L. Foster, PhD U.S. Geological Survey GMEG Menlo Park, CA Arsenic is an element of concern in mined gold deposits around the world Spenceville (Cu-Au-Ag) Lava Cap (Nevada) Ketza River (Au) Empire Mine (Nevada) low-sulfide, qtz Au sulfide and oxide ore Argonaut Mine (Au) bodies Don Pedro Harvard/Jamestown Ruth Mine (Cu) Kelly/Rand (Au/Ag) Goldenville, Caribou, and Montague (Au) The common arsenic-rich particles in hard-rock gold mines have long been known Primary Secondary Secondary/Tertiary Iron oxyhydroxide (“rust”) “arsenian” pyrite containing arsenic up to 20 wt% -1 Scorodite FeAsO 2H O As pyrite Fe(As,S)2 4 2 Kankite : FeAsO4•3.5H2O Reich and Becker (2006): maximum of 6% As-1 Arseniosiderite Ca2Fe3(AsO4)3O2·3H2O Arsenopyrite FeAsS Jarosite KFe3(SO4)2(OH)6 Yukonite Ca7Fe12(AsO4)10(OH)20•15H2O Tooleite [Fe6(AsO3)4(SO4)(OH)4•4H2O Pharmacosiderite KFe (AsO ) (OH) •6–7H O arsenide n- 4 4 3 4 2 n = 1-3 But it is still difficult to predict with an acceptable degree of uncertainty which forms will be present • thermodynamic data lacking or unreliable for many important phases • kinetic barriers to equilibrium • changing geochemical conditions (tailings management) Langmuir et al. (2006) GCA v70 Lava Cap Mine Superfund Site, Nevada Cty, CA Typical exposure pathways at arsenic-contaminated sites are linked to particles and their dissolution in aqueous fluids ingestion of arsenic-bearing water dissolution near-neutral, low -

Arsenic and Lead Contamination in Oklahoma Soils Arsenic and Lead Are Naturally Occurring Elements Present in Rocks
Arsenic and Lead Contamination in Oklahoma Soils Arsenic and lead are naturally occurring elements present in rocks. As these rocks erode, arsenic and lead get in soils, waters and plants. The United States Geological Survey (USGS) reports an average of 7.4 parts per million (ppm) as the arsenic concentration in soils for the entire United States and 7.2 ppm as the average arsenic concentration in soils for the western U.S.(1) Background arsenic concentrations in Central Oklahoma soils range from 0.6 to 21 ppm.(2) The USGS reports an average lead concentration of 13 parts per million (ppm) in soils for central Oklahoma(2) and an average lead concentration of 18 ppm for the Western United States.(3) Pollution from historic lead and zinc smelters in Oklahoma left residues of lead, arsenic and cadmium in area soils. Contamination was also spread by using smelter debris as fill material, driveways and roads. These former smelters were primarily located in eastern Oklahoma. Some arsenic contamination above naturally occurring levels is also attributed to agricultural, energy and industrial practices. For example, arsenic-containing insecticides and herbicides were widely used on vegetables, fruits and field crops from the 1900s to around 1950. Coal combustion, wood preserving and smelting operations are known to be sources of arsenic contamination.(1) Lead was once commonly used in paint and in plumbing. Lead was an additive to gasoline for many years and has also been found in ceramics, mini-blinds and other products. Homes built before 1970 commonly have lead-based paint. Renovation activities and peeling or flaking paint can release lead inside the home. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

United States Patent Office
Patented Mar. 12, 1946 2,396.465 UNITED STATES PATENT OFFICE PREPARATION OF SODUMARSENTE Erroll Hay Karr, Tacoma, Wash, assignor to The Pennsylvania Salt Manufacturing Company, Philadelphia, Pa, a corporation of Pennsyl vania No Drawing. Application October 12, 1943, Serial No. 506,00 3 Claims. (C. 23-53) This invention relates to the production of metal arsenites from alkali metal hydroxide and high arsenic content water soluble, alkali metal arsenic trioxide without the necessity of using arsenites, and more particularly, it relates to an the alkali metal hydroxide in powder form. improved method of causing a controlled reac Another object of the invention is to provide tion between an alkali metal hydroxide and a cheap and easily performed process for the arsenic trioxide to obtain a granular product in preparation of alkali metal arsenites from con a one-step operation. centrated solutions of caustic alkali and pow It is known that solid sodium arsenite can be dered arsenic trioxide in standard mixing equip prepared by mixing together powdered Solid ment. caustic soda and arsenic oxide in a heap and O An additional object of the invention is to pro initiating the reaction by adding a small quan vide a process for obtaining alkali metal arsenites tity of water. Ullmann's Encyclopedia der Techn. in granular form from concentrated solutions of Chemie., volume I, page 588. This method will caustic alkali and powdered arsenic trioxide. yield a solid material but it is attended by sev A further object of the invention is to provide eral disadvantages. 5 a process for the preparation of granular alkali . -

Detection, Distribution and Health Risk Assessment of Toxic Heavy
foods Article Detection, Distribution and Health Risk Assessment of Toxic Heavy Metals/Metalloids, Arsenic, Cadmium, and Lead in Goat Carcasses Processed for Human Consumption in South-Eastern Nigeria Emmanuel O. Njoga 1,* , Ekene V. Ezenduka 1 , Chiazor G. Ogbodo 1, Chukwuka U. Ogbonna 2 , Ishmael F. Jaja 3 , Anthony C. Ofomatah 4 and Charles Odilichukwu R. Okpala 5,* 1 Department of Veterinary Public Health and Preventive Medicine, Faculty of Veterinary Medicine, University of Nigeria, Nsukka 410001, Nigeria; [email protected] (E.V.E.); [email protected] (C.G.O.) 2 Department of Biochemistry, Federal University of Agriculture Abeokuta, Ogun State 110124, Nigeria; [email protected] 3 Department of Livestock and Pasture Science, University of Fort Hare, Alice 5700, South Africa; [email protected] 4 National Centre for Energy Research and Development, University of Nigeria, Nsukka 410001, Nigeria; [email protected] 5 Department of Functional Food Products Development, Faculty of Biotechnology and Food Science, Wrocław University of Environmental and Life Sciences, 51-630 Wrocław, Poland Citation: Njoga, E.O.; Ezenduka, * Correspondence: [email protected] (E.O.N.); [email protected] (C.O.R.O.) E.V.; Ogbodo, C.G.; Ogbonna, C.U.; Jaja, I.F.; Ofomatah, A.C.; Okpala, Abstract: Notwithstanding the increased toxic heavy metals/metalloids (THMs) accumulation in C.O.R. Detection, Distribution and (edible) organs owed to goat0s feeding habit and anthropogenic activities, the chevon remains Health Risk Assessment of Toxic increasingly relished as a special delicacy in Nigeria. Specific to the South-Eastern region, however, Heavy Metals/Metalloids, Arsenic, Cadmium, and Lead in Goat there is paucity of relevant data regarding the prevalence of THMs in goat carcasses processed for Carcasses Processed for Human human consumption. -

High-Entropy Alloy: Challenges and Prospects
Materials Today Volume 19, Number 6 July/August 2016 RESEARCH Review High-entropy alloy: challenges and prospects RESEARCH: Y.F. Ye, Q. Wang, J. Lu, C.T. Liu and Y. Yang* Centre for Advanced Structural Materials, Department of Mechanical and Biomedical Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Kowloon, Hong Kong High-entropy alloys (HEAs) are presently of great research interest in materials science and engineering. Unlike conventional alloys, which contain one and rarely two base elements, HEAs comprise multiple principal elements, with the possible number of HEA compositions extending considerably more than conventional alloys. With the advent of HEAs, fundamental issues that challenge the proposed theories, models, and methods for conventional alloys also emerge. Here, we provide a critical review of the recent studies aiming to address the fundamental issues related to phase formation in HEAs. In addition, novel properties of HEAs are also discussed, such as their excellent specific strength, superior mechanical performance at high temperatures, exceptional ductility and fracture toughness at cryogenic temperatures, superparamagnetism, and superconductivity. Due to their considerable structural and functional potential as well as richness of design, HEAs are promising candidates for new applications, which warrants further studies. Introduction When designing alloys, researchers previously focused on the From ancient times, human civilization has striven to develop new corners of a phase diagram to develop a conventional alloy, which materials [1], discovering new metals and inventing new alloys occupy only a small portion of the design space, as illustrated by that have played a pivotal role for more than thousands of years. -

Open Phd Thesis - Final Draft.Pdf
The Pennsylvania State University The Graduate School College of Earth and Mineral Sciences DEPOSITION, CHARACTERIZATION, AND THERMOMECHANICAL FATIGUE OF NICKEL ALUMINIDE AND RUTHENIUM ALUMINIDE THIN FILMS A Dissertation in Materials Science and Engineering by Jane A. Howell © 2010 Jane A. Howell Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2010 The dissertation of Jane A. Howell was reviewed and approved* by the following: Suzanne E. Mohney Professor of Materials Science and Engineering Dissertation Co-Advisor Co-Chair of Committee Christopher L. Muhlstein Associate Professor of Materials Science and Engineering Dissertation Co-Advisor Co-Chair of Committee Joseph R. Flemish Senior Scientist and Professor of Materials Science and Engineering Clifford J. Lissenden Professor of Engineering Science and Mechanics Joan M. Redwing Professor of Materials Science and Engineering Chair, Intercollege Graduate Degree Program in Materials Science and Engineering *Signatures are on file in the Graduate School ii ABSTRACT Intermetallic thin films have properties that make them attractive for applications such as metallizations, high temperature coatings, microelectromechanical systems, and diffusion barrier layers. In this study B2 aluminide films (NiAl and RuAl) have been deposited and characterized. Both intermetallics could be deposited at temperatures near room temperature using co- sputtering with an as-deposited resistivity of 45.5 ± 1.5 μΩcm for NiAl and 157 ± 4 μΩcm for RuAl. Ni/Al multilayers with a wavelength of 30 nm and below were fully reacted to form NiAl after annealing for 2 h at 400°C. These films had a resistivity of 15.5-26.7 μΩcm (wavelengths from 15.4-30 nm) after a 4 h anneal at 400°C, and the lower values of resistivity correspond to films with larger wavelengths. -

Structure and Properties of Cast Ti-Al-Si Alloys
materials Article Structure and Properties of Cast Ti-Al-Si Alloys Anna Knaislová 1 , Pavel Novák 1,* , Jiˇrí Linhart 1, Ivo Szurman 2, KateˇrinaSkotnicová 2 , Jan Juˇrica 2 and Tomáš Ceganˇ 2 1 Department of Metals and Corrosion Engineering, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic; [email protected] (A.K.); [email protected] (J.L.) 2 Department of Non-Ferrous Metals, Refining and Recycling, Faculty of Materials Science and Technology, VSB—Technical University of Ostrava, 17. listopadu 15, 708 33 Ostrava-Poruba, Czech Republic; [email protected] (I.S.); [email protected] (K.S.); [email protected] (J.J.); [email protected] (T.C.)ˇ * Correspondence: [email protected] Abstract: Intermetallic compounds based on Ti-Al- (Si) are attractive materials with good thermal stability and low density. However, the production of these materials is quite complicated. Partially modified conventional methods of melting metallurgy are most often used due to availability, possible high productivity, and relatively low production costs. Therefore, some technologies for the production of intermetallics based on Ti-Al are currently available, but with certain disadvantages, which are caused by poor casting properties or extreme reactivity of the melt with crucibles. Some shortcomings can be eliminated by modifying the melting technology, which contributes to increasing the cost of the process. The work deals with the preparation of Ti-Al-Si intermetallic compounds with different contents of aluminum and silicon, which were produced by centrifugal casting in an induction vacuum furnace Linn Supercast-Titan. This process could contribute to the commercial use of these alloys in the future. -

Are You Living in an Area with Mine Tailings? Arsenic and Health Did You Know?
Are you living in an area with mine tailings? Arsenic and health Did you know? • Mine tailings from gold mining often contain high levels of arsenic. • Arsenic in mine tailings may be harmful to health. – Most people have only a very small chance of being affected. – Babies and young children are more likely to be affected than adults. – Children who eat small handfuls of mine tailings are especially at risk – they may even get arsenic poisoning. • Unlike historical mines, licensed gold mines today are highly regulated, including the management of tailings. • If you live near historical mine tailings, you can reduce the risk to your health by reducing the amount of soil and dust that you swallow. • Fruit and vegetables that are grown on mine tailings may absorb arsenic. – Eating fruit and vegetables with raised levels of arsenic may sometimes be harmful to health. Many towns and cities in Victoria have been built in areas with a history of gold mining. Mine tailings that contain arsenic are spread over large areas of land, including land now used for housing. This booklet contains information for people who live in these areas. It gives information on what you need to know and actions you can take to protect your family’s health. b What is arsenic? Arsenic is a substance that is found naturally in rock, often near gold deposits. It was used as a poison to kill insects that attack animals, timber, fruit and vegetables. In some situations, arsenic causes health effects in people. What are mine tailings and why do they contain high levels of arsenic? When gold is mined, rocks are brought to the surface and crushed to extract the gold. -

Possible Health Risks from Exposure to Arsenic, Lead, and Polycyclic
How can I reduce my family’s exposure to arsenic, lead, and PAHs in surface soil? • Wash pets often. • Wash children’s toys often. Separate indoor and outdoor toys. Possible Health Risks from Exposure to Arsenic, • Wash children’s hands and feet after they have been playing outside. Lead, and Polycyclic Aromatic Hydrocarbons • Do not eat food, chew gum, or smoke when working in the yard. • Parents monitor their children’s behavior while playing outdoors and prevent their children from eating soil, or unintentionally swallowing 35th Avenue Site Public Health Evaluation, North Birmingham, it. A covered sandbox can reduce children’s digging in soil in other Alabama areas of the yard. Introduction Are the fruits and vegetables from my garden safe to eat? The United States Environmental Protection Agency (US EPA) sampled • PAHs were not found in garden produce. Exposure to PAHs in surface soil from over 1,100 residential properties and homegrown produce should not cause health problems. garden produce from a few area gardens at the 35th Avenue site. The • The arsenic and lead levels found in garden produce are low. Eating Agency for Toxic Substances and Disease Registry (ATSDR) looked at garden produce should not cause health problems if this is the only the arsenic, lead, and polycyclic aromatic hydrocarbons (called PAHs) way people are exposed to arsenic or lead. However, when people levels to see if people’s contact with these compounds could harm are also exposed to high levels of arsenic and lead in soil, or other ways, they are more likely to develop their health. -

Effect of Si on Fe-Rich Intermetallic Formation and Mechanical Properties of Heat- Treated Al-Cu-Mn-Fe Alloys Yuliang Zhao1, 2
Effect of Si on Fe-rich intermetallic formation and mechanical properties of heat- treated Al-Cu-Mn-Fe alloys Yuliang Zhao1, 2, Weiwen Zhang1*, Chao Yang1, Datong Zhang1, Zhi Wang1 1National Engineering Research Center of Near-net-shape Forming for Metallic Materials, South China University of Technology, Guangzhou, 510641, China 2School of Engineering & Computer Science, University of Hull, East Yorkshire, HU6 7RX, UK *Corresponding author. Tel: +86-20-87112022, Fax: +86-20-87112111. E-mail: [email protected] Abstract: The effect of Si on Fe-rich intermetallics formation and mechanical properties of heat-treated squeeze cast Al-5.0Cu-0.6Mn-0.7Fe alloy was investigated. Our results show that increasing Si content promotes the formation of Al15(FeMn)3(SiCu)2 (α-Fe), and varying the morphology of T (Al20Cu3Mn2) where the size decreases and the amount increases. The major reason is that Si promotes heterogeneous nucleation of the intermetallics leading to finer precipitates. Si addition significantly enhances ultimate tensile strength and yield strength of the alloys. The strengthening effect is mainly owing to the dispersoid strengthening by increasing volume fraction of T phase and less harmful α-Fe with a compact structure, which make the cracks more difficult to initiate and propagation during tensile test. The squeeze cast Al-5.0Cu-0.6Mn-0.7Fe alloy with 1.1% Si shows significantly improved mechanical properties than the alloy without Si addition, which has tensile strength of 386 MPa, yield strength of 280 MPa and elongation of 8.6%. Key Words: Al; casting; microstructure 1 I. Introduction Al-Cu alloys have been widely used in automobile manufacturing, space technology and aerospace industry owing to their high specific strength, good heat resistance and excellent fatigue properties [1-3].