Dissolution of Uranyl Microprecipitates in Subsurface Sediments at Hanford Site, USA
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Uraninite Alteration in an Oxidizing Environment and Its Relevance to the Disposal of Spent Nuclear Fuel
TECHNICAL REPORT 91-15 Uraninite alteration in an oxidizing environment and its relevance to the disposal of spent nuclear fuel Robert Finch, Rodney Ewing Department of Geology, University of New Mexico December 1990 SVENSK KÄRNBRÄNSLEHANTERING AB SWEDISH NUCLEAR FUEL AND WASTE MANAGEMENT CO BOX 5864 S-102 48 STOCKHOLM TEL 08-665 28 00 TELEX 13108 SKB S TELEFAX 08-661 57 19 original contains color illustrations URANINITE ALTERATION IN AN OXIDIZING ENVIRONMENT AND ITS RELEVANCE TO THE DISPOSAL OF SPENT NUCLEAR FUEL Robert Finch, Rodney Ewing Department of Geology, University of New Mexico December 1990 This report concerns a study which was conducted for SKB. The conclusions and viewpoints presented in the report are those of the author (s) and do not necessarily coincide with those of the client. Information on SKB technical reports from 1977-1978 (TR 121), 1979 (TR 79-28), 1980 (TR 80-26), 1981 (TR 81-17), 1982 (TR 82-28), 1983 (TR 83-77), 1984 (TR 85-01), 1985 (TR 85-20), 1986 (TR 86-31), 1987 (TR 87-33), 1988 (TR 88-32) and 1989 (TR 89-40) is available through SKB. URANINITE ALTERATION IN AN OXIDIZING ENVIRONMENT AND ITS RELEVANCE TO THE DISPOSAL OF SPENT NUCLEAR FUEL Robert Finch Rodney Ewing Department of Geology University of New Mexico Submitted to Svensk Kämbränslehantering AB (SKB) December 21,1990 ABSTRACT Uraninite is a natural analogue for spent nuclear fuel because of similarities in structure (both are fluorite structure types) and chemistry (both are nominally UOJ. Effective assessment of the long-term behavior of spent fuel in a geologic repository requires a knowledge of the corrosion products produced in that environment. -

United States Department of the Interior Geological Survey
UNITED STATES DEPARTMENT OF THE INTERIOR GEOLOGICAL SURVEY A CATALOG OF STUDY MATERIAL OF RADIOACTIVE MINERALS T. W. Stern October 1950 Trace Elements Investigations Report 129 JAN 1 0 2081' CONTENTS Page Introduction ......................... Group A. Uranium and thorium minerals Group B. Minerals with minor amounts of uranium and thorium ................................ 62 Group C. Synthetic uranium compounds pre pared by the U. S. Geological Survey ............... ^9 A CATALOG OF STUDY MATERIAL OF RADIOACTIVE MINERALS by T. W. Stern Introduction This catalog is a list of specimens of radioactive minerals for which the Geological Survey has been collecting optical, X-ray dif fraction, and other data. The specimens are in the study collections of the U. S. National Museum, the Mineralogical Museum of Harvard University (including some specimens on loan at Harvard from the American Museum of Natural History), and the Geological Survey. The listing does not include all specimens in these institutions, but it does include those specimens for which X-ray patterns can be made available as well as representative specimens from significant local ities. In addition, references are given to material on deposit, at the U. S. National Museum, that has been described in the literature. Because of the rarity of much of the material here listed, re quests for study material must be considered individually. Regardless of the quantity of material, any request granted by the U. S. National Museum will have to be accepted on the terms laid down by that organ ization to conserve limited or rare minerals. Those minerals that show external crystal form but do not give X-ray powder diffraction patterns are listed with the word "ignited" after the X-ray powder diffraction photograph number. -

Thn Auertcan M Rlueralocrsr
THn AUERTcANM rluERALocrsr JOURNAL OF TIIE MINDRALOGICAL SOCIETY OF ANIERICA vbl.41 JULY-AUGUST, 1956 Nos. 7 and 8 MTNERAL COMPOSTTTON OF G'UMMTTE*f Crrllonl FnoNonr, H artard Llniaersity,Cambrid,ge, M ass., and. U. S. GeologicalSurwy, Washington, D.C. ABSTRACT The name gummite has been wideiy used for more than 100 years as a generic term to designate fine-grained yellow to orange-red alteration products of uraninite whose true identity is unknown. A study of about 100 specimens of gummite from world-wide localities has been made by r-ray, optical, and chemical methods. rt proved possible to identify almost all of the specimens with already known uranium minerals. Gummite typicalty occurs as an alteration product of uraninite crystals in pegmatite. Such specimensshow a characteristic sequenceof alteration products: (1) A central core of black or brownish-black uraninite. (2) A surrounding zone, yellow to orange-red, composed chiefly of hydrated lead uranyl oxides. This zone constitutes the traditional gummite. It is principally composed of fourmarierite, vandendriesscheite and two unidentified phases (Mineral -4 and Mineral c). Less common constituents are clarkeite, becquerelite, curite, and schoepite. (3) An outer silicate zone. This usually is dense with a greenish-yellow color and is composed of uranophane or beta-uranophane; it is sometimes soft and earthy with a straw-yellow to pale-brown color and is then usually composed of kasolite or an unidenti- fied phase (Minerat B). Soddyite and sklodowskite occur rarely. There are minor variations in the above general sequence. rt some specimens the core may be orange-red gummite without residual uraninite or the original uraninite crystal may be wholly converted to silicates. -

Follow up the Leaching Efficiency of Uranium Series from High-Grade Granite Sample with High Concentration of Sulfuric Acid
PROCEEDINGS: Nada et al. Follow up the leaching efficiency of uranium series from high-grade granite sample with high concentration of sulfuric acid A. Nada1, N. Imam2*, A. Ghanem1 1 Physics Department, Faculty of Women for Art, Science and Education, Ain Shams University, 11757, Egypt. 2 National Institute of Oceanography and Fisheries, Cairo, 11516, Egypt Keywords: Leaching efficiency, Granite sample, Sulfuric acid Presenting author, e-mail: [email protected] Introduction Materials and methods Uranium is the most representative actinide element that Sample preparation is of fundamental importance in the nuclear fuel cycle. The red granite sample from Gabal Gattar, the Northern Uranium is a naturally occurring radionuclide which has Eastern Desert (2352.8 ppm), was crushed and grinded 238 9 235 three isotopes ( U (t1/2=4.5x10 yr) 99.72 %, U into 63 mesh and then quartered to obtain representative 8 234 5 (t1/2=7.1x10 yr) 0.0055 % and U (t1/2=2.5x10 yr) sample. The mineralogical characterization of the granite 0.72 %. It has several oxidation states tetravalent and sample was done using X-Ray Diffraction (XRD) (Model hexavalent which dominant in the environment. D8 discover manufacture by USA). The sample is Hexavalent uranium is more dissoluble and mobile than measured by y-spectrometry, using an HPGe-detector to tetravalent uranium. Extraction of uranium is indeed determine the activity concentrations (Bq) of Uranium a hydrometallurgical operation in which uranium is isotopes (238U, 235U, and 234U), 230Th and 226Ra series. The directly leached first by suitable acid or alkaline reagents conditions of the leaching procedures were 50 g of sample, (Kraiz et al., 2016). -

SIMPLE URANIUM OXIDES, HYDROXIDES U4+ + U6+, SIMPLE and COMPLEX URANYL HYDROXIDES in ORES Andrey A
New Data on Minerals. 2011. Vol. 46 71 SIMPLE URANIUM OXIDES, HYDROXIDES U4+ + U6+, SIMPLE AND COMPLEX URANYL HYDROXIDES IN ORES Andrey A. Chernikov Fersman Mineralogical Museum, Russian Academy of Sciences, Moscow, [email protected], [email protected] The review of published and new own data of simple uranium oxides revealed that the formation of five simple oxides is probable: nasturan, sooty pitchblende, uraninite, uranothorianite, and cerianite. Among simple oxides, nasturan, sooty pitchblende, and uraninite are the most abundant in ores varied in genesis and mineralogy. Uranothorianite or thorium uraninite (aldanite) is occasional in the ores, while cerianite is believed in U-P deposits of Northern Kazakhstan. Hydrated nasturan is the most abundant among three uranium (IV+VI) hydroxides in uranium ores. Insignificant ianthinite was found in few deposits, whereas cleusonite was indentified only in one deposit. Simple uranyl hydroxides, schoepite, metaschoepite, and paraschoepite, are widespread in the oxidized ores of the near-surface part of the Schinkolobwe deposit. They are less frequent at the deeper levels and other deposits. Studtite and metastudtite are of insignificant industrial importance, but are of great inter- est to establish genesis of mineral assemblages in which they are observed, because they are typical of strongly oxidized conditions of formation of mineral assemblages and ores. The X-ray amorphous urhite associated with hydrated nasturan and the X-ray amorphous hydrated matter containing ferric iron and U6+ described for the first time at the Lastochka deposit, Khabarovsk krai, Russia are sufficiently abundant uranyl hydroxides in the oxidized uranium ores. Significant complex uranyl hydroxides with interlayer K, Na, Ca, Ba, Cu, Pb, and Bi were found basically at a few deposits: Schinkolobwe, Margnac, Wölsendorf, Sernyi, and Tulukuevo, and are less frequent at the other deposits, where quite large monomineralic segregations of nasturan and crystals of uraninite were identified. -
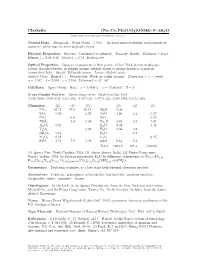
Clarkeite (Na, Ca, Pb)(UO2)O(OH) • 0−1H2O C 2001-2005 Mineral Data Publishing, Version 1
Clarkeite (Na, Ca, Pb)(UO2)O(OH) • 0−1H2O c 2001-2005 Mineral Data Publishing, version 1 Crystal Data: Hexagonal. Point Group: 32/m. As dense microcrystalline replacements of uraninite, which may be mineralogically zoned. Physical Properties: Fracture: Conchoidal to splintery. Tenacity: Brittle. Hardness = 4–4.5 D(meas.) = 6.29–6.39 D(calc.) = 6.74 Radioactive. Optical Properties: Opaque, transparent in thin grains. Color: Dark brown, mahogany- brown, chocolate-brown, or reddish orange; reddish brown to orange-brown or orange in transmitted light. Streak: Yellowish brown. Luster: Slightly waxy. Optical Class: Biaxial (–). Pleochroism: Weak; in darker oranges. Dispersion: r< v,weak. α = 1.997 β = 2.098 γ = 2.108 2V(meas.) = 30◦–50◦ Cell Data: Space Group: R3m. a = 3.954(1) c = 17.660(3) Z = 3 X-ray Powder Pattern: Fanny Gouge mine, North Carolina, USA. 3.195 (100), 5.903 (64), 3.365 (56), 2.707 (34), 1.9772 (33), 2.945 (28), 1.6410 (23) Chemistry: (1) (2) (3) (1) (2) (3) UO3 81.72 79.9 83.14 MgO 0.28 SiO2 0.50 0.03 CaO 2.84 2.1 0.37 UO2 0.8 SrO 0.29 ThO2 2.4 0.49 Na2O 3.44 2.3 7.01 Al2O3 0.92 K2O 0.48 0.42 + Y2O3 0.82 H2O 3.36 4.2 − RE2O3 2.62 H2O 1.3 Fe2O3 0.18 H2O [3.47] PbO 3.71 7.9 3.96 insol. 0.14 0.2 Total 100.19 101.1 [100.00] (1) Spruce Pine, North Carolina, USA. -

STRONG and WEAK INTERLAYER INTERACTIONS of TWO-DIMENSIONAL MATERIALS and THEIR ASSEMBLIES Tyler William Farnsworth a Dissertati
STRONG AND WEAK INTERLAYER INTERACTIONS OF TWO-DIMENSIONAL MATERIALS AND THEIR ASSEMBLIES Tyler William Farnsworth A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry. Chapel Hill 2018 Approved by: Scott C. Warren James F. Cahoon Wei You Joanna M. Atkin Matthew K. Brennaman © 2018 Tyler William Farnsworth ALL RIGHTS RESERVED ii ABSTRACT Tyler William Farnsworth: Strong and weak interlayer interactions of two-dimensional materials and their assemblies (Under the direction of Scott C. Warren) The ability to control the properties of a macroscopic material through systematic modification of its component parts is a central theme in materials science. This concept is exemplified by the assembly of quantum dots into 3D solids, but the application of similar design principles to other quantum-confined systems, namely 2D materials, remains largely unexplored. Here I demonstrate that solution-processed 2D semiconductors retain their quantum-confined properties even when assembled into electrically conductive, thick films. Structural investigations show how this behavior is caused by turbostratic disorder and interlayer adsorbates, which weaken interlayer interactions and allow access to a quantum- confined but electronically coupled state. I generalize these findings to use a variety of 2D building blocks to create electrically conductive 3D solids with virtually any band gap. I next introduce a strategy for discovering new 2D materials. Previous efforts to identify novel 2D materials were limited to van der Waals layered materials, but I demonstrate that layered crystals with strong interlayer interactions can be exfoliated into few-layer or monolayer materials. -

Clarkeite: New Chemical and Structural Data
American Mineralogist, Volume 82, pages 607±619, 1997 Clarkeite: New chemical and structural data ROBERT J. FINCH* AND RODNEY C. EWING Department of Earth and Planetary Sciences, University of New Mexico, Albuquerque, New Mexico 87131, U.S.A. ABSTRACT Clarkeite crystallizes during metasomatic replacement of pegmatitic uraninite by late- stage, oxidizing hydrothermal ¯uids. Samples are zoned compositionally: Clarkeite, which is Na rich, surrounds a K-rich core (commonly with remnant uraninite) and is surrounded by more Ca-rich material; volumetrically, clarkeite is most abundant. Clarkeite is hexag- onal (space group Rm3Å )a53.954(4), c 5 17.73(1) AÊ (Z 5 3). The structure of clarkeite is based on anionic sheets of the form [(UO2)(O,OH)2]. The sheets are bonded to each other through interlayer cations and H2O molecules. The empirical formula for clarkeite from the Fanny Gouge mine near Spruce Pine, North Carolina, is: {Na0.733K0.029Ca0.021Sr0.009Y0.024Th0.006Pb0.058}S0.880[(UO2)0.942O0.918(OH)1.082](H2O)0.069. Na predominates and the Pb is radiogenic. The general formula for clarkeite is 21 31 41 {(Na,K)pMMMPbqr s x}[(UO2)1 2 xO1 2 y(OH)1 1 y](H2O)z where Na .. K and p . (q 1 r 1 s). The number of O22 ions and OH groups in the structural unit is determined by the net charge of the interlayer cations (except Pb): y 5 1 2 (p 1 2q 1 3r 1 4s). This suggests that the ideal formula for ideal end-member clarkeite is Na[(UO2)O(OH)](H2O)0-1. -

The Pegmatite Dyke at Rømteland
The pegmatite dyke at Rømteland. A c/e^ci/ii/on of the mme^a/F and a ck/FcuFFlon of the mineral paragenesis, especially within the system (Fe, Mn)O-TiO2-Y2O3-(Nb, Ta)205-Si02 By Thor L. Sverdrup Contents lntro^uction 125 Magnetite 161 125 162 Description of the area 127 162 6etelM!NstionB 128 163 History 129 Orangite 163 Description of the pegmatite .... 130 Palygorskite 164 Temperature of formation of the Plagioclase 164 pegmatite 134 p^rite 166 Magmatic origin; supporting Quartz 166 odBervation3 142 3amalBliite 167 OeBci'lption ot tlie mineral 143 Spessartite 168 143 3pkene 169 147 169 153 170 Apatite 156 170 Bastnåsite 156 Uraninite 170 Lei-yl 157 Clarkeite 171 Liotite 157 171 Oalcite 158 l<2B3olite 171 Chalcopyrite 158 a-uranophane 171 158 ?ircon 171 Chlorite (Daphnite) 158 Mineral paragenesis at Rømteland 173 Columbite 158 The two adjacent four component 159 system with the oxides: (Fe, Luxenite 159 Mn)O-Y2O3-(Nb, Ta)2O5-TiOé> ?luolite 160 SiO2 181 Nematite 160 161 Sammendrag 191 Ilmenite 161 195 Introduction. The pegmatite described is located at Rømteland, about 7 km W of Vigeland, Vest-Agder county, southern Norway. Field investigations were made in the summers of 1955 and 1956, and v the laboratory work was done under the direction of Professor Dr. Tom F. W. Barth and Dr. H. Neumann. The pe^matite is BUllouncie6 by a hornblende bearing quartz monzonite which Barth (1945) has called farsundite. Major (1939) believes the quartz monzonite to be a magmatic rock which during its emplacement has torn loose pieces of the sur rounding gneiss and partly assimilated these. -

Complete Title of My Thesis
GEOCHEMISTRY OF URANIUM AT MINERAL-WATER INTERFACES: RATES OF SORPTION-DESORPTION AND DISSOLUTION-PRECIPITATION REACTIONS Thesis by Daniel Giammar In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy California Institute of Technology Pasadena, California 2001 (Defended May 8, 2001) ii © 2001 Daniel Giammar All Rights Reserved iii Acknowledgments My advisor Janet Hering has supported and encouraged me throughout my graduate student career. From her I have learned a great deal about aquatic chemistry and, by her example, much about scientific curiosity and discipline. She is consistently my best and most thorough critic. Michael Hoffmann’s encouragement and suggestions have contributed in many ways to my work. George Rossman’s mineralogical insight led to some of the most interesting experiments in this work. Jim Morgan has been a mentor and role model throughout my study at Caltech, and I would particularly like to thank him for his emphasis on the fundamentals, both scientifically and on the basketball court. I would like to acknowledge the profound influence that Susan Leach, my middle school life and earth sciences teacher, had on my decision to pursue a career in environmental science and engineering. David Dzombak at Carnegie Mellon encouraged me to go to graduate school and remains a great role model for me. At Caltech, Gerald Wasserburg’s enthusiasm for geochemistry got me interested in uranium and his encouragement contributed to the initiation of this work. The summer undergraduate research projects of Helen Claudio and Yi-Ping Liu constitute portions of this work. When I would run into a problem with the X-ray diffractometer or scanning electron microscope, Chi Ma was always there to bail me out. -

Attachment SOTERM)
Title 40 CFR Part 191 Subparts B and C Compliance Recertification Application for the Waste Isolation Pilot Plant Appendix SOTERM-2009 Actinide Chemistry Source Term United States Department of Energy Waste Isolation Pilot Plant Carlsbad Field Office Carlsbad, New Mexico Appendix SOTERM-2009 Actinide Chemistry Source Term Title 40 CFR Part 191 Subparts B and C Compliance Recertification Application 2009 Table of Contents SOTERM-1.0 Introduction ............................................................................................SOTERM-1 SOTERM-2.0 Expected WIPP Repository Conditions, Chemistry, and Processes ......SOTERM-3 SOTERM-2.1 Ambient Geochemical Conditions .....................................................SOTERM-3 SOTERM-2.2 Repository Conditions........................................................................SOTERM-3 SOTERM-2.2.1 Repository Pressure.....................................................................SOTERM-5 SOTERM-2.2.2 Repository Temperature..............................................................SOTERM-5 SOTERM-2.2.3 Water Content and Relative Humidity ........................................SOTERM-5 SOTERM-2.2.4 Minimum Repository Brine Volume...........................................SOTERM-6 SOTERM-2.2.5 DRZ.............................................................................................SOTERM-7 SOTERM-2.3 Repository Chemistry.........................................................................SOTERM-7 SOTERM-2.3.1 WIPP Brine..................................................................................SOTERM-7 -

Minerals Localities
MINERALS and their LOCALITIES This book is respectfully dedicated to the memory of Dr. John Sinkankas for his kind initiative and support to publish this book in English version. MINERALS and their LOCALITIES Jan H. Bernard and Jaroslav Hyršl Edited by Vandall T. King © 2004 by Granit, s.r.o. © 2004 Text by Jan H. Bernard and Jaroslav Hyršl © 2004 Photos by Jaroslav Hyršl (463), Studio Granit (534), Jaromír Tvrdý (34), Petr Zajíček (4) The photographed specimens are from the collections of both authors as well as from many other collections. The autors are grateful to all institutions and persons who allowed to photograph their specimens for this book. Front cover photos: Turquoise, polished, 55 mm, Zhilandy, Kazakhstan, G Galena, 45 mm, Madan, Bulgaria, G Sphalerite, xx 12 mm, Morococha, Peru, H Gypsum, xx 40 mm, Las Salinas, Peru, H Variscite, xx 5 mm, Itumbiara, Brazil, H Rhodochrosite, polished, 50 mm, Capillitas, Argentina, H Back cover photo: Wolframite, 45 mm, Yaogangxian, China, H Page 1: Muscovite, 45 mm, Linopolis, Brazil, H Page 2: Vivianite, 100 mm, Huanzala, Bolivia, H Page 3: Liddicoatite, polished, 70 mm, Anjanabonoina, Madagaskar, G Page 5: Opal - fire, polished, 50 mm, Mezezo, Ethiopia, G Page 12: Brazilianite, 35 mm, Linopolis, Brazil, H Page 13: Gold, 35 mm, Eagle's Nest Mine, California, G Published by Granit, s.r.o. Štefánikova 43, 150 00 Praha 5, Czech Republic e-mail: [email protected] www.granit-publishing.cz Composition and reproduction by Studio VVG, Prague Printed in Czech Republic by Finidr, s.r.o., Český Těšín 14/02/03/01 All rights reserved.