A Molecular Phylogeny of the Lythraceae and Inference of the Evolution of Heterostyly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

To View Fulltext
A CONTRIBUTION TO THE EMBRYOLOGY OF SONNERATIACEAE. BY JILLELLA VENKATESWARLU. (From the Department of Botany, Benares Hi~,du U~iz,ersity, Benares.) Received April 14, 1937. (Communicated by iV[r. A. C. Joshi, M.sc.) SONNERATIACEm iS a small family of the Myrtifloreae. According to Engler and Prantl (1898, 1908), it includes four genera, Sonneratia, Duabanga, Xenodendron and Crypteronia, comprising about a dozen species. All these are found in the tropics, the monotypic genus Xenodendron being confin6d to New Guinea and the rest being mostly restricted to the Indo-Malayan region. Hutchinson (1926) in his recent system of classification has sepa- rated the genus Crypteronia into a separate family Crypteroniacem and the family Sonneratiaeem as defined by him includes only three genera. Bc:ntham a!~d I~ooker (1%2-67), on the other hand, include these genera in the family Lythracem, to which there is little doubt that these are very closely related. As the writer had been recently studying the embryology of the family Lythracem (Joshi and Venkateswarln, 1935 a, 1935 b, 1935 c, 1936), it was thought desirable to study the family Sonneratiacem also from the comparative point of view and to see how far its embryological features agree with those of the Lythracem. The previous work on this family is limited to an investigation on Sonneratia apetala Linn. by Karsten (1891). tIis observations, however, are very fragmentary and also partly erroneous as pointed out by me in preliminary notes (Venkateswarlu, 1936a, 1936b) relating to the plants described in the present paper. The present paper deals with the two chief genera of the family, namely, Duabanga and Sonneratia. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -

Scarlet Ammannia (Ammannia Robusta) in Canada
PROPOSED Species at Risk Act Recovery Strategy Series Adopted under Section 44 of SARA Recovery Strategy for the Scarlet Ammannia (Ammannia robusta) in Canada Scarlet Ammannia 2014 Recommended citation: Environment Canada. 2014. Recovery Strategy for the Scarlet Ammannia (Ammannia robusta) in Canada [Proposed]. Species at Risk Act Recovery Strategy Series. Environment Canada, Ottawa. XXI pp. + Appendix. For copies of the recovery strategy, or for additional information on species at risk, including the Committee on the Status of Endangered Wildlife in Canada (COSEWIC) Status Reports, residence descriptions, action plans, and other related recovery documents, please visit the Species at Risk (SAR) Public Registry (www.sararegistry.gc.ca). Cover illustration: © Emmet J. Judziewicz Également disponible en français sous le titre « Programme de rétablissement de l’ammannie robuste (Ammannia robusta) au Canada [Proposition] » © Her Majesty the Queen in Right of Canada, represented by the Minister of the Environment, 2014. All rights reserved. ISBN Catalogue no. Content (excluding the illustrations) may be used without permission, with appropriate credit to the source. RECOVERY STRATEGY FOR THE SCARLET AMMANNIA (Ammannia robusta) IN CANADA 2014 Under the Accord for the Protection of Species at Risk (1996), the federal, provincial, and territorial governments agreed to work together on legislation, programs, and policies to protect wildlife species at risk throughout Canada. In the spirit of cooperation of the Accord, the Government of British Columbia has given permission to the Government of Canada to adopt the “Recovery Strategy for the scarlet ammannia (Ammannia robusta) in British Columbia and Ontario” (Part 2) under Section 44 of the Species at Risk Act (SARA). -

Flora.Sa.Gov.Au/Jabg
JOURNAL of the ADELAIDE BOTANIC GARDENS AN OPEN ACCESS JOURNAL FOR AUSTRALIAN SYSTEMATIC BOTANY flora.sa.gov.au/jabg Published by the STATE HERBARIUM OF SOUTH AUSTRALIA on behalf of the BOARD OF THE BOTANIC GARDENS AND STATE HERBARIUM © Board of the Botanic Gardens and State Herbarium, Adelaide, South Australia © Department of Environment, Water and Natural Resources, Government of South Australia All rights reserved State Herbarium of South Australia PO Box 2732 Kent Town SA 5071 Australia J. Adelaide Bot. Gard. 1(1) 55-59 (1976) A SUMMARY OF THE FAMILY LYTHRACEAE IN THE NORTHERN TERRITORY (WITH ADDITIONAL COMMENTS ON AUSTRALIAN MATERIAL) by A. S. Mitchell Arid Zone Research Institute, Animal Industry and Agriculture Branch, Department of the Northern Territory, Alice Springs, N.T. 5750. Abstract This paper presents a synopsis of the nomenclature of the family Lythraceae in the Northern Territory. Keysto the genera and species have been prepared. The family Lythraceae has been neglected in Australian systematics, andas a result both the taxonomy and nomenclature are confused. Not since the early work of Koehne (1881, 1903) has there been any major revision of the family. Recent work has been restricted to regional floras (Polatschek and Rechinger 1968; Chamberlain 1972; Dar 1975), with Bentham's Flora (1886) being the most recenton the family in Australia. From a survey of the available literature the author has attempted to extract all the relevant names applicable to Australian material and to present them solelyas a survey of the nomenclature of the group. No type material has beenseen, and the only material examined was that lodged in the Department of the Northern Territory Herbariaat Alice Springs (NT) and Darwin (DNA). -

Diversity of Wisconsin Rosids
Diversity of Wisconsin Rosids . oaks, birches, evening primroses . a major group of the woody plants (trees/shrubs) present at your sites The Wind Pollinated Trees • Alternate leaved tree families • Wind pollinated with ament/catkin inflorescences • Nut fruits = 1 seeded, unilocular, indehiscent (example - acorn) *Juglandaceae - walnut family Well known family containing walnuts, hickories, and pecans Only 7 genera and ca. 50 species worldwide, with only 2 genera and 4 species in Wisconsin Carya ovata Juglans cinera shagbark hickory Butternut, white walnut *Juglandaceae - walnut family Leaves pinnately compound, alternate (walnuts have smallest leaflets at tip) Leaves often aromatic from resinous peltate glands; allelopathic to other plants Carya ovata Juglans cinera shagbark hickory Butternut, white walnut *Juglandaceae - walnut family The chambered pith in center of young stems in Juglans (walnuts) separates it from un- chambered pith in Carya (hickories) Juglans regia English walnut *Juglandaceae - walnut family Trees are monoecious Wind pollinated Female flower Male inflorescence Juglans nigra Black walnut *Juglandaceae - walnut family Male flowers apetalous and arranged in pendulous (drooping) catkins or aments on last year’s woody growth Calyx small; each flower with a bract CA 3-6 CO 0 A 3-∞ G 0 Juglans cinera Butternut, white walnut *Juglandaceae - walnut family Female flowers apetalous and terminal Calyx cup-shaped and persistant; 2 stigma feathery; bracted CA (4) CO 0 A 0 G (2-3) Juglans cinera Juglans nigra Butternut, white -

ISTA List of Stabilized Plant Names 7Th Edition
ISTA List of Stabilized Plant Names th 7 Edition ISTA Nomenclature Committee Chair: Dr. M. Schori Published by All rights reserved. No part of this publication may be The Internation Seed Testing Association (ISTA) reproduced, stored in any retrieval system or transmitted Zürichstr. 50, CH-8303 Bassersdorf, Switzerland in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior ©2020 International Seed Testing Association (ISTA) permission in writing from ISTA. ISBN 978-3-906549-77-4 ISTA List of Stabilized Plant Names 1st Edition 1966 ISTA Nomenclature Committee Chair: Prof P. A. Linehan 2nd Edition 1983 ISTA Nomenclature Committee Chair: Dr. H. Pirson 3rd Edition 1988 ISTA Nomenclature Committee Chair: Dr. W. A. Brandenburg 4th Edition 2001 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 5th Edition 2007 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 6th Edition 2013 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 7th Edition 2019 ISTA Nomenclature Committee Chair: Dr. M. Schori 2 7th Edition ISTA List of Stabilized Plant Names Content Preface .......................................................................................................................................................... 4 Acknowledgements ....................................................................................................................................... 6 Symbols and Abbreviations .......................................................................................................................... -

Wood Anatomy of Lythraceae Assigned to The
Ada Bot. Neerl. 28 (2/3), May 1979, p.117-155. Wood anatomy of the Lythraceae P. Baas and R.C.V.J. Zweypfenning Rijksherbarium, Leiden, The Netherlands SUMMARY The wood anatomy of 18 genera belonging to the Lythraceae is described. The diversity in wood structure of extant Lythraceae is hypothesized to be derived from a prototype with scanty para- I tracheal parenchyma, heterogeneous uniseriate and multiseriate rays, (septate)libriform fibres with minutely bordered pits, and vessels with simple perforations. These characters still prevail in a number of has been limited in of Lythraceae. Specialization very most Lythraceae shrubby or herbaceous habit: these have juvenilistic rays composed mainly of erect rays and sometimes com- pletely lack axial parenchyma. Ray specialization towards predominantly uniseriate homogeneous concomitant with fibre abundant and with rays, dimorphism leading to parenchyma differentiation, the advent of chambered crystalliferous fibres has been traced in the “series” Ginoria , Pehria, Lawsonia , Physocalymma and Lagerstroemia. The latter genus has the most specialized wood anatomy in the family and has species with abundant parenchyma aswell as species with alternating fibres. with its bands of dimorphous septate Pemphis represents an independent specialization vasicentric parenchyma and thick-walled nonseptate fibres. The affinities of with other are discussed. Pun Psiloxylon, Lythraceae Myrtales ica, Rhynchocalyx , Oliniaceae,Alzatea, Sonneratiaceae, Onagraceae and Melastomataceae all resemble Lythraceae in former accommodated in the without their wood anatomy. The three genera could even be family its wood anatomical Alzatea and Sonneratia differ in minor details extending range. Oliniaceae, only from order facilitate identification of wood tentative the Lythraceae. In to samples, keys to genera or groups ofgenera of Lythraceae as well as to some species of Lagerstroemiaare presented. -

A Review of the Plants of the Princeton Chert (Eocene, British Columbia, Canada)
Botany A review of the plants of the Princeton chert (Eocene, British Columbia, Canada) Journal: Botany Manuscript ID cjb-2016-0079.R1 Manuscript Type: Review Date Submitted by the Author: 07-Jun-2016 Complete List of Authors: Pigg, Kathleen; Arizona State University, School of Life Sciences DeVore, Melanie; Georgia College and State University, Department of Biological &Draft Environmental Sciences Allenby Formation, Aquatic plants, Fossil monocots, Okanagan Highlands, Keyword: Permineralized floras https://mc06.manuscriptcentral.com/botany-pubs Page 1 of 75 Botany A review of the plants of the Princeton chert (Eocene, British Columbia, Canada) Kathleen B. Pigg , School of Life Sciences, Arizona State University, PO Box 874501, Tempe, AZ 85287-4501, USA Melanie L. DeVore , Department of Biological & Environmental Sciences, Georgia College & State University, 135 Herty Hall, Milledgeville, GA 31061 USA Corresponding author: Kathleen B. Pigg (email: [email protected]) Draft 1 https://mc06.manuscriptcentral.com/botany-pubs Botany Page 2 of 75 A review of the plants of the Princeton chert (Eocene, British Columbia, Canada) Kathleen B. Pigg and Melanie L. DeVore Abstract The Princeton chert is one of the most completely studied permineralized floras of the Paleogene. Remains of over 30 plant taxa have been described in detail, along with a diverse assemblage of fungi that document a variety of ecological interactions with plants. As a flora of the Okanagan Highlands, the Princeton chert plants are an assemblage of higher elevation taxa of the latest early to early middle Eocene, with some components similar to those in the relatedDraft compression floras. However, like the well known floras of Clarno, Appian Way, the London Clay, and Messel, the Princeton chert provides an additional dimension of internal structure. -

New Records and Notes on the Plant Diversity of Singapore
Gardens' Bulletin Singapore 71 (2): 401–406. 2019 401 doi: 10.26492/gbs71(2).2019-07 Flora of Singapore precursors, 16: New records and notes on the plant diversity of Singapore L.M.J. Chen1, B.E.E. Duyfjes2, Ali Ibrahim1 & W.J.J.O. de Wilde2 1Singapore Botanic Gardens, National Parks Board, 1 Cluny Road, 259569 Singapore 2 Naturalis Biodiversity Center, section Botany, P.O. Box 9517, 2300 RA Leiden, The Netherlands [email protected] ABSTRACT. Due to ongoing work for the Flora of Singapore, a new family record for Singapore, the Stemonaceae, with one species, Stemona curtisii Hook.f., is recorded. In addition, Ammannia crassicaulis Guill. & Perr. in the Lythraceae is newly recorded as naturalising in Singapore. Notes on two rare species, Hernandia nymphaeifolia (C.Presl) Kubitzki in the Hernandiaceae and Securidaca philippinensis Chodat in the Polygalaceae, currently being revised for the Flora of Singapore are presented. Keywords. Aquarium plant, aquatic plant, freshwater swamp forest, naturalised species New Records of Stemonaceae and Lythraceae 1. Stemona curtisii Hook.f., Fl. Brit. India 6: 298 (1892). – TYPE: Peninsular Malaysia, Penang, Curtis 1522 (lectotype K [K000292133], designated here). Perennial twiner with fasciculate tubers, plant 1–2 m high, glabrous, somewhat branched. Leaves alternate, seldom opposite; petiole 4–12 cm long; lamina sometimes slightly rough, (narrowly) ovate, 6–21 × 2.5–12.5 cm, base (shallowly or) broadly cordate, apex acuminate, 11–17(–19)-palmately-veined. Inflorescences 1–many- flowered; peduncle 1–10(–18) cm long; bracts 6–20 mm long; pedicel 10–20 mm long. Flowers tepals narrowly triangular, 17–30 × 5(–7) mm; stamens 18–25 mm long, filaments c. -

Interim Report GVG.Pdf
Disclaimer: Due to COVID-19, the team could not carry forward the work as planned. The remaining work will be carried out in the coming field season i.e., from September 2020-August 2021 (depending on the pandemic situation). The information provided here are preliminary findings and no final inferences are drawn at this preliminary stage. Further data collection and analysis are needed for drawing final inferences. Adhikarimayum, A.S., Ahmad, A., Deepan, C., Deshwal, D., Himanshu, C.L., Lamgouhao, N., and Gopi, G.V. (2020). Status and conservation of tigers and their habitats in hill ranges of northeast India with special reference to Mishmi hills, Manipur and Nagaland. First interim project progress report. Technical report. Wildlife Institute of India. TR/2020/12. i | P a g e TABLE OF CONTENTS Page no Acknowledgements iii Executive Summary 1 Introduction 4 Objectives 5 CHAPTERS CHAPTER 1 Kamlang Tiger Reserve 6 CHAPTER 2 Mehao Wildlife Sanctuary 26 CHAPTER 3 Dibang Wildlife Sanctuary 47 CHAPTER 4 Manipur and Nagaland 69 APPENDICES Appendix A MoU between WII and NTCA Appendix B Permission letter from Government of Arunachal Pradesh Appendix C Permission letter from Government of Manipur Appendix D Permission letter from Government of Nagaland ii | P a g e Acknowledgements First and foremost, we would like to thank the people of Dibang Valley, Lower Dibang Valley, Lohit districts in Arunachal Pradesh, Peren district in Nagaland, Tamenglong and Senapati districts in Manipur for their generous support and help during our fieldwork. We are immensely grateful to Dr. Debabrata Swain and Dr. Anup K Nayak, former Member Secretaries of NTCA, Dr.S.P.Yadav, Member Secretary, NTCA, Mr. -

Phylogenetic Analysis of the Sonneratiaceae and Its Relationship to Lythraceae Based on ITS Sequences of Nrdna
_______________________________________________________________________________www.paper.edu.cn J. Plant Res. 113: 253-258, 2000 Journal of Plant Research 0 by The Botanical Society of JaDan 2000 Phylogenetic Analysis of the Sonneratiaceae and its Relationship to Lythraceae Based on ITS Sequences of nrDNA Suhua Shi’*, Yelin Huang’, Fengxiao Tan’, Xingjin He’ and David E. Boufford’ School of Life Sciences, Zhongshan University, Guangzhou 510275, China Harvard University Herbaria, 22 Divinity Avenue, Cambridge, MA 02138-2020,U.S. A. Phylogenetic analyses were conducted using seven Asia. Species of Sonneratia are trees of mangrove swamps species in two genera of Sonneratiaceae, seven species in and seacoasts generally, and the inland genus Duabanga is five genera of the related Lythraceae and 2 outgroups an evergreen component of the rainforest belt (Backer et a/. based on DNA sequences of the internal transcribed spacer 1954). Approximate five to 11 species are recognized in the (ITS) and the 5.8s coding region of the nuclear ribosomal family worldwide, seven of which occur from south central to DNA to determine the proper systematic placement of the southeast China (Backer and Steenis 1951, Duck and Jackes Sonneratiaceae. Paraphyly of the traditional Lythraceae 1987, KO 1983). The mangrove genus Sonneratia occurs was shown with the genus Lagerstmemia nested within the mainly on Hainan Island in China and consists of five native Sonneratiaceae. The Sonneratiaceae occurred within the species, S. a/& Smith, S. caseolaris (L.) Engler, S, hainanensis Lythraceae with high bootstrap value support (96%). The W.C. KO, E.Y. Chen & W.Y. Chen, S. ovata Backer, S. par- two traditional genera constituting Sonneratiaceae were in acaseolaris W.C. -
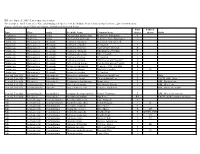
Threatened and Endangered Species List
Effective April 15, 2009 - List is subject to revision For a complete list of Tennessee's Rare and Endangered Species, visit the Natural Areas website at http://tennessee.gov/environment/na/ Aquatic and Semi-aquatic Plants and Aquatic Animals with Protected Status State Federal Type Class Order Scientific Name Common Name Status Status Habit Amphibian Amphibia Anura Gyrinophilus gulolineatus Berry Cave Salamander T Amphibian Amphibia Anura Gyrinophilus palleucus Tennessee Cave Salamander T Crustacean Malacostraca Decapoda Cambarus bouchardi Big South Fork Crayfish E Crustacean Malacostraca Decapoda Cambarus cymatilis A Crayfish E Crustacean Malacostraca Decapoda Cambarus deweesae Valley Flame Crayfish E Crustacean Malacostraca Decapoda Cambarus extraneus Chickamauga Crayfish T Crustacean Malacostraca Decapoda Cambarus obeyensis Obey Crayfish T Crustacean Malacostraca Decapoda Cambarus pristinus A Crayfish E Crustacean Malacostraca Decapoda Cambarus williami "Brawley's Fork Crayfish" E Crustacean Malacostraca Decapoda Fallicambarus hortoni Hatchie Burrowing Crayfish E Crustacean Malocostraca Decapoda Orconectes incomptus Tennessee Cave Crayfish E Crustacean Malocostraca Decapoda Orconectes shoupi Nashville Crayfish E LE Crustacean Malocostraca Decapoda Orconectes wrighti A Crayfish E Fern and Fern Ally Filicopsida Polypodiales Dryopteris carthusiana Spinulose Shield Fern T Bogs Fern and Fern Ally Filicopsida Polypodiales Dryopteris cristata Crested Shield-Fern T FACW, OBL, Bogs Fern and Fern Ally Filicopsida Polypodiales Trichomanes boschianum