Mil U Ottawa
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Insights from the Web Usage Statistics in Plos Article-Level Metrics
CORE Metadata, citation and similar papers at core.ac.uk Provided by PubMed Central The Spread of Scientific Information: Insights from the Web Usage Statistics in PLoS Article-Level Metrics Koon-Kiu Yan1,2, Mark Gerstein1,2,3* 1 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, United States of America, 2 Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut, United States of America, 3 Department of Computer Science, Yale University, New Haven, Connecticut, United States of America Abstract The presence of web-based communities is a distinctive signature of Web 2.0. The web-based feature means that information propagation within each community is highly facilitated, promoting complex collective dynamics in view of information exchange. In this work, we focus on a community of scientists and study, in particular, how the awareness of a scientific paper is spread. Our work is based on the web usage statistics obtained from the PLoS Article Level Metrics dataset compiled by PLoS. The cumulative number of HTML views was found to follow a long tail distribution which is reasonably well-fitted by a lognormal one. We modeled the diffusion of information by a random multiplicative process, and thus extracted the rates of information spread at different stages after the publication of a paper. We found that the spread of information displays two distinct decay regimes: a rapid downfall in the first month after publication, and a gradual power law decay afterwards. We identified these two regimes with two distinct driving processes: a short-term behavior driven by the fame of a paper, and a long-term behavior consistent with citation statistics. -

Media Kit 2006-2007 Plos Biology
PLoS Biology our flagship title * 2.4 million pages fully downloaded per year * 1.5 million visits per year * 100,000 unique visitors per month Media Kit 2006-2007 www.PLoS.org table OF CONTENTS 1 LETTER FROM THE PUBLISHER 2 A LITTLE HISTORY 3 EXPANDED ADVERTISING OPPORTUNITIES 4 HEADLINE FACTS AND FIGURES 5 COOL ACCOLADES 6 RATES & SPONSORSHIP PACKAGES 7 PREMIUM AD TYPES 8 TECHNICAL SPECIFICATIONS 9 CLOSING DATES 10 GENERAL INFORMATION PLoS Media Kit 2006-2007 i [email protected] (415) 568 3446 1a 1b 1c letter from the publisher redesigned web sites give advertisers more choice and impact Dear PLoS Advertiser, Welcome to PLoS. As a premier open access publisher, our journals have rapidly become “must reads” for the world’s most innovative minds. Infl uential researchers throughout the world choose PLoS because they know that scientifi c progress occurs faster when results are freely accessible to all. This is what makes them ardent fans of open access and compels them to publish their work in PLoS journals. The PLoS fl agship journals, PLoS Biology and PLoS Medicine, are already established as top-tier publications with impact factors of 14.7 and 8.4, respectively. All our newer titles have also quickly gained prominence amongst the leaders in their fi elds. Later in 2006, we will launching PLoS ONE, another groundbreaking publishing initiative. By advertising with PLoS, your products and services will reach well-connected and successful individuals with the purchasing power and authority to give your brand power. Plus, you will also be associating your name with a positive force for change and the most exciting projects in scientifi c and medical publishing. -

Finding What Works in Health Care: Standards for Systematic Reviews
This PDF is available from The National Academies Press at http://www.nap.edu/catalog.php?record_id=13059 Finding What Works in Health Care: Standards for Systematic Reviews ISBN Jill Eden, Laura Levit, Alfred Berg, and Sally Morton, Editors; Committee 978-0-309-16425-2 on Standards for Systematic Reviews of Comparative Effectiveness Research; Institute of Medicine 372 pages 6 x 9 PAPERBACK (2011) Visit the National Academies Press online and register for... Instant access to free PDF downloads of titles from the NATIONAL ACADEMY OF SCIENCES NATIONAL ACADEMY OF ENGINEERING INSTITUTE OF MEDICINE NATIONAL RESEARCH COUNCIL 10% off print titles Custom notification of new releases in your field of interest Special offers and discounts Distribution, posting, or copying of this PDF is strictly prohibited without written permission of the National Academies Press. Unless otherwise indicated, all materials in this PDF are copyrighted by the National Academy of Sciences. Request reprint permission for this book Copyright © National Academy of Sciences. All rights reserved. Finding What Works in Health Care: Standards for Systematic Reviews Committee on Standards for Systematic Reviews of Comparative Effectiveness Research Board on Health Care Services Jill Eden, Laura Levit, Alfred Berg, and Sally Morton, Editors THE NATIONAL ACADEMIES PRESS Washington, D.C. www.nap.edu Copyright © National Academy of Sciences. All rights reserved. Finding What Works in Health Care: Standards for Systematic Reviews THE NATIONAL ACADEMIES PRESS 500 Fifth Street, N.W. Washington, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. -

Open Source Biology: a Means to Address the Access & Research Gaps
Santa Clara High Technology Law Journal Volume 23 | Issue 2 Article 2 2007 Open Source Biology: A Means to Address the Access & Research Gaps Katherine M. Nolan-Stevaux Follow this and additional works at: http://digitalcommons.law.scu.edu/chtlj Part of the Law Commons Recommended Citation Katherine M. Nolan-Stevaux, Open Source Biology: A Means to Address the Access & Research Gaps, 23 Santa Clara High Tech. L.J. 271 (2006). Available at: http://digitalcommons.law.scu.edu/chtlj/vol23/iss2/2 This Article is brought to you for free and open access by the Journals at Santa Clara Law Digital Commons. It has been accepted for inclusion in Santa Clara High Technology Law Journal by an authorized administrator of Santa Clara Law Digital Commons. For more information, please contact [email protected]. OPEN SOURCE BIOLOGY: A MEANS TO ADDRESS THE ACCESS & RESEARCH GAPS? Katherine M. Nolan-Stevauxt Abstract Although Americans enjoy access to a wide range of drugs to treat all types of diseases, ranging from life-threatening to life- sustaining, numerous serious illnesses exist for which either there are no drugs available or worse the drugs that do exist are not available in a particular marketplace, such as the developing world. This articlefocuses on how an open source licensing system premised on patent law can foster drug development to benefit the developing world. The first section discusses the access and research gaps and explains how patents, in part, contribute to these gaps. The second section briefly explains open source licensing practices in software before discussing why an open source approach attracts biomedical researchers and how it might differ from open source approaches in software. -

Research Data Curation and Management Bibliography
Research Data Curation and Management Bibliography Charles W. Bailey Jr. Research Data Curation and Management Bibliography Copyright © 2021 by Charles W. Bailey, Jr. This work is licensed under a Creative Commons Attribution 4.0 International License, https://creativecommons.org/licenses/by/4.0/. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA. Original cover image before modification by Liam Huang. That image is under a Creative Commons Attribution 2.0 Generic License (https://creativecommons.org/licenses/by/2.0/). Digital Scholarship, Houston, TX. http://www.digital-scholarship.org/ The author makes no warranty of any kind, either express or implied, for information in the Research Data Curation and Management Bibliography, which is provided on an "as is" basis. The author does not assume and hereby disclaims any liability to any party for any loss or damage resulting from the use of information in the Research Data Curation and Management Bibliography. Cite as: Bailey, Charles W., Jr. Research Data Curation and Management Bibliography. Houston: Digital Scholarship, 2021. http://www.digital- scholarship.org/rdcmb/rdcmb.htm. Preface The Research Data Curation and Management Bibliography includes over 800 selected English-language articles and books that are useful in understanding the curation of digital research data in academic and other research institutions. The "digital curation" concept is still evolving. In "Digital Curation and Trusted Repositories: Steps toward Success," Christopher A. Lee and Helen R. Tibbo define digital curation as follows: Digital curation involves selection and appraisal by creators and archivists; evolving provision of intellectual access; redundant storage; data transformations; and, for some materials, a commitment to long-term preservation. -
Status, Use and Impact of Sharing Individual Participant Data from Clinical Trials: a Scoping Review
Open access Original research BMJ Open: first published as 10.1136/bmjopen-2021-049228 on 18 August 2021. Downloaded from Status, use and impact of sharing individual participant data from clinical trials: a scoping review Christian Ohmann ,1 David Moher ,2 Maximilian Siebert ,3 Edith Motschall ,4 Florian Naudet 5 To cite: Ohmann C, Moher D, ABSTRACT Strengths and limitations of this study Siebert M, et al. Status, Objectives To explore the impact of data- sharing use and impact of sharing initiatives on the intent to share data, on actual data ► Exhaustive review of both the literature and the main individual participant sharing, on the use of shared data and on research output data from clinical trials: a initiatives in data sharing. and impact of shared data. scoping review. BMJ Open ► Analysis of the full data- sharing process covering in- Eligibility criteria All studies investigating data- sharing 2021;11:e049228. doi:10.1136/ tention to share, actual sharing, use of shared data, practices for individual participant data (IPD) from clinical bmjopen-2021-049228 research output and impact. trials. ► Retrieval and synthesis of information proved to be ► Prepublication history and Sources of evidence We searched the Medline database, difficult because of a very siloed landscape where additional supplemental material the Cochrane Library, the Science Citation Index Expanded for this paper are available each initiative/platform operates with its own and the Social Sciences Citation Index via Web of Science, online. To view these files, metrics. and preprints and proceedings of the International please visit the journal online. ► Data sharing is a moving target in a rapidly changing Congress on Peer Review and Scientific Publication. -
The Spread of Scientific Information: Insights from the Web Usage Statistics in Plos Article-Level Metrics
The Spread of Scientific Information: Insights from the Web Usage Statistics in PLoS Article-Level Metrics Koon-Kiu Yan1,2, Mark Gerstein1,2,3* 1 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, United States of America, 2 Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut, United States of America, 3 Department of Computer Science, Yale University, New Haven, Connecticut, United States of America Abstract The presence of web-based communities is a distinctive signature of Web 2.0. The web-based feature means that information propagation within each community is highly facilitated, promoting complex collective dynamics in view of information exchange. In this work, we focus on a community of scientists and study, in particular, how the awareness of a scientific paper is spread. Our work is based on the web usage statistics obtained from the PLoS Article Level Metrics dataset compiled by PLoS. The cumulative number of HTML views was found to follow a long tail distribution which is reasonably well-fitted by a lognormal one. We modeled the diffusion of information by a random multiplicative process, and thus extracted the rates of information spread at different stages after the publication of a paper. We found that the spread of information displays two distinct decay regimes: a rapid downfall in the first month after publication, and a gradual power law decay afterwards. We identified these two regimes with two distinct driving processes: a short-term behavior driven by the fame of a paper, and a long-term behavior consistent with citation statistics. -
Solr Vignette
Local setup of Solr and querying using solr R package, on Mac OSX A general purpose R interface to Solr This package only deals with exracting data from a Solr endpoint, not writing data (pull request or holla if you’re interested in writing solr data). Solr info • Solr home page • Highlighting help • Faceting help • Installing Solr on Mac using homebrew • Install and Setup SOLR in OSX, including running Solr Quick start Install Install dependencies install.packages(c("rjson", "plyr", "httr", "XML", "assertthat")) Install solr install.packages("devtools") library(devtools) install_github("ropensci/solr") library(solr) Define stuff Your base url and a key (if needed). This example should work. You do need to pass a key to the Public Library of Science search API, but it apparently doesn’t need to be a real one. url <- "http://api.plos.org/search" key <- "key" Search solr_search(q = "*:*", rows = 2, fl = "id", base = url, key = key) ## http://api.plos.org/search?q=*:*&start=0&rows=2&wt=json&fl=id ## id ## 1 10.1371/journal.pone.0060627 ## 2 10.1371/journal.pbio.0000080 Facet 1 solr_facet(q = "*:*", facet.field = "journal", facet.query = c("cell", "bird"), base = url, key = key) ## http://api.plos.org/search?q=*:*&facet.query=cell&facet.query=bird&facet.field=journal&key=key&wt=json&fl=DOES_NOT_EXIST&facet=true ## $facet_queries ## term value ## 1 cell 85941 ## 2 bird 8588 ## ## $facet_fields ## $facet_fields$journal ## X1 X2 ## 1 plos one 742824 ## 2 plos genetics 35463 ## 3 plos pathogens 31152 ## 4 plos computational biology 26016 ## 5 -
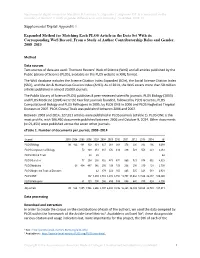
Supplemental Digital Appendix 1 Expanded Method for Matching
Supplemental digital content for Macaluso B, Larivière V, Sugimoto T, Sugimoto CR. Is science built on the shoulders of women? A study of gender differences in contributorship. Acad Med. 2016; 91. Supplemental Digital Appendix 1 Expanded Method for Matching Each PLOS Article in the Data Set With its Corresponding WoS Record, From a Study of Author Contributorship Roles and Gender, 2008–2013 Method Data sources Two sources of data are used: Thomson Reuters’ Web of Science (WoS) and all articles published by the Public Library of Science (PLOS), available on the PLOS website in XML format. The WoS database includes the Science Citation Index Expanded (SCIe), the Social Science Citation Index (SSCI), and the Art & Humanities Citation Index (AHCI). As of 2014, the WoS covers more than 50 million articles published in almost 20,000 journals. The Public Library of Science (PLOS) publishes 8 peer-reviewed scientific journals. PLOS Biology (2003) and PLOS Medicine (2004) were the two first journals founded, followed by PLOS Genetics, PLOS Computational Biology and PLOS Pathogens in 2005, by PLOS ONE in 2006 and PLOS Neglected Tropical Diseases in 2007. PLOS Clinical Trials was published between 2006 and 2007. Between 2003 and 2014, 127,911 articles were published in PLOS journals (eTable 1). PLOS ONE is the most prolific, with 106,460 documents published between 2006 and October 9, 2014. Other documents (n=21,451) were published across the seven other journals. eTable 1. Number of documents per journal, 2003–2014 Journal 2003 2004 2005 2006 2007 -

Artemisia Annua, Artemisinin, Acts and Malaria Control in Africa: the Interplay of Tradition, Science and Public Policy
1 Working Paper September 20, 2010 Artemisia annua, Artemisinin, ACTs and Malaria Control in Africa: The Interplay of Tradition, Science and Public Policy Dana G. Dalrymple 1 As to diseases, make a habit of two things –to help, or at least to do no harm. - Hippocrates, 460-377 BC2 The ends of this branch of knowledge [chemical philosophy] are the applications of natural substances to new uses, for increasing the comforts and enjoyments of man… - Sir Humphry Davy, 1812 It would be foolhardy of anyone to predict when and how malaria will be conquered. - Socrates Litsios, 1996 Abstract The key ingredient in the leading treatment for malaria in Africa - artemisinin - comes not from high- tech research, but is an extract of an ancient medicinal plant, Artemisia annua, commonly known as Artemisia. Chloroquine and replacement drugs have lost effectiveness with the development of resistance and have increasingly been replaced by derivatives of artemisinin combined with other drugs. Known as artemisinin–based combination therapies (ACTs), they provide the most effective treatment at present. This has led to efforts to increase cultivated production of Artemisia in the short run and to develop, through biological and chemical research, synthetic substitutes in the longer run. The resulting juxtaposition of activities and players provides both opportunities and challenges for society. While individual components have been examined, there is little in the way of comprehensive analysis. This paper attempts to weave the many complex and dynamic components - historical, scientific, technical, economic - together in order to aid understanding of the issues and facilitate development of informed public/private policies and actions. -
Chapter Thirteen
The Basement Interviews Freeing the scientific literature Harold Varmus, Nobel laureate, former director of the US National Institutes of Health, and co-founder of open access publisher Public Library of Science, talks to Richard Poynder. Published on June 5th 20061 Harold Varmus was born in 1939, on the south shore of Long Island, New York, a product, as he put it in an autobiographical note he wrote years later,2 of "the early twentieth century emigration of Eastern European Jewry to New York City and its environs." His paternal grandfather, Jacob Varmus, had arrived in the New World from a small village near Warsaw just after the turn of the century, settling first as a farmer in Newburgh, New York, and subsequently working as a hatter in Newark, New Jersey. His paternal grandmother, Eleanor, was a victim of the influenza epidemic of 1918, dying when his father was eleven. Varmus' maternal grandparents — Harry and Regina Barasch — came originally from farming villages around Linz, Austria. Settling in Freeport, New York, they ran a children's clothing store. Three years before Varmus' birth, his parents also settled in Freeport, where his father — who had trained as a doctor — established a general medical practice. From there Varmus' mother, Beatrice, commuted to a social services job in New York City. A lake with alligators When the United States entered the War, Varmus' father was assigned to an air force hospital near Winter Park, Florida, where the family resided until 1946. "My first memories," Varmus recalled later, "were to be of long beaches, and bass fishing on a lake with alligators." It was in Florida that Varmus' only sibling, Ellen Jane, was born. -

2021 Program Book
Department of Family and Community Medicine National Center for Medical Education Development and Research The 5th Annual Communities of Practice Conference The Future of Medical Education and Clinical Services Transformation: Meeting the Needs of Vulnerable Populations in 2030 May 26-27, 2021 · A Virtual Conference Department of Family and Community Medicine National Center for Medical Education Development and Research The 5th Annual Communities of Practice Conference The Future of Medical Education and Clinical Services Transformation: Meeting the Needs of Vulnerable Populations in 2030 May 26-27, 2021 · A Virtual Conference What is a Community of Practice? 4th Annual Communities of Practice Conference A community of practice (CoP) is a group of people who share a craft or a profession. The concept was first proposed by cognitive anthropologist Jean Lave and educational theorist Etienne Wenger in their 1991 book Situated Learning (Lave & Wenger 1991). Wenger then significantly expanded on the concept in his 1998 book Communities of Practice (Wenger 1998). A CoP can evolve naturally because of the members’ common interest in a particular domain or area, or it can be created deliberately with the goal of gaining knowledge related to a specific field. It is through the process of sharing information and experiences with the group that members learn from each other, and have an opportunity to develop personally and professionally (Lave & Wenger 1991). (Lave & Wenger 1991). The Primary Care Training and Enhancement Academic Units (AU) project is supported in part through a cooperative agreement (UH1HP30348) with the US Department of Health and Human Services (DHHS)/Health Resources and Services Administration (HRSA) and Department of Family and Community Medicine, School of Medicine, Meharry Medical College.