Tepzz 59Z6¥4B T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Mechanisms of Action of Ppars
12 Dec 2001 8:6 AR AR149-24.tex AR149-24.SGM LaTeX2e(2001/05/10) P1: GSR Annu. Rev. Med. 2002. 53:409–35 Copyright c 2002 by Annual Reviews. All rights reserved THE MECHANISMS OF ACTION OF PPARS Joel Berger and David E. Moller Department of Molecular Endocrinology, Merck Research Laboratories, P.O. Box 2000, Rahway, New Jersey 07065; e-mail: joel [email protected]; david [email protected] Key Words PPAR, nuclear receptors, diabetes, dyslipidemia ■ Abstract The peroxisome proliferator-activated receptors (PPARs) are a group of three nuclear receptor isoforms, PPAR ,PPAR, and PPAR, encoded by different genes. PPARs are ligand-regulated transcription factors that control gene expression by binding to specific response elements (PPREs) within promoters. PPARs bind as heterodimers with a retinoid X receptor and, upon binding agonist, interact with co- factors such that the rate of transcription initiation is increased. The PPARs play a critical physiological role as lipid sensors and regulators of lipid metabolism. Fatty acids and eicosanoids have been identified as natural ligands for the PPARs. More potent synthetic PPAR ligands, including the fibrates and thiazolidinediones, have proven effective in the treatment of dyslipidemia and diabetes. Use of such ligands has allowed researchers to unveil many potential roles for the PPARs in pathological states including atherosclerosis, inflammation, cancer, infertility, and demyelination. Here, we present the current state of knowledge regarding the molecular mechanisms of PPAR action and the involvement of the PPARs in the etiology and treatment of several chronic diseases. INTRODUCTION The peroxisome proliferator-activated receptors (PPARs) form a subfamily of the nuclear receptor superfamily. -

Treatment Patterns, Persistence and Adherence Rates in Patients with Type 2 Diabetes Mellitus in Japan: a Claims- Based Cohort Study
Open access Research BMJ Open: first published as 10.1136/bmjopen-2018-025806 on 1 March 2019. Downloaded from Treatment patterns, persistence and adherence rates in patients with type 2 diabetes mellitus in Japan: a claims- based cohort study Rimei Nishimura,1 Haruka Kato,2 Koichi Kisanuki,2 Akinori Oh,2 Shinzo Hiroi,2 Yoshie Onishi,3 Florent Guelfucci,4 Yukio Shimasaki2 To cite: Nishimura R, Kato H, ABSTRACT Strengths and limitations of this study Kisanuki K, et al. Treatment Objective To determine real-world trends in antidiabetic patterns, persistence and drug use, and persistence and adherence, in Japanese ► This retrospective evaluation of administrative adherence rates in patients patients with type 2 diabetes mellitus (T2DM). with type 2 diabetes mellitus claims data (2011–2015) using the Japan Medical Design Retrospective evaluation of administrative claims in Japan: a claims-based Data Center (JMDC) and Medical Data Vision (MDV) data (2011–2015) using the Japan Medical Data Center cohort study. BMJ Open databases was conducted to determine real-world (JMDC) and Medical Data Vision (MDV) databases. 2019;9:e025806. doi:10.1136/ trends in antidiabetic drug use, and persistence and Setting Analysis of two administrative claims databases bmjopen-2018-025806 adherence, in Japanese patients with type 2 dia- for Japanese patients with T2DM. betes mellitus (T2DM); 40 908 and 90 421 patients ► Prepublication history and Participants Adults (aged ≥18 years) with an International additional material for this were included from the JMDC and MDV databases, Classification of Diseases, 10th Revision code of T2DM and paper are available online. To respectively. at least one antidiabetic drug prescription. -

CDR Clinical Review Report for Soliqua
CADTH COMMON DRUG REVIEW Clinical Review Report Insulin glargine and lixisenatide injection (Soliqua) (Sanofi-Aventis) Indication: adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus inadequately controlled on basal insulin (less than 60 units daily) alone or in combination with metformin. Service Line: CADTH Common Drug Review Version: Final (with redactions) Publication Date: January 2019 Report Length: 118 Pages Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services. While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. -

Download Product Insert (PDF)
PRODUCT INFORMATION Remogliflozin A Item No. 14340 CAS Registry No.: 329045-45-6 OH Formal Name: 5-methyl-4-[[4-(1-methylethoxy) N O OH phenyl]methyl]-1-(1-methylethyl)- N 1H-pyrazol-3-yl β-D- O glucopyranoside OH Synonym: GSK189074 OH MF: C23H34N2O7 FW: 450.5 Purity: ≥98% λ: 229 nm UV/Vis.: max O Supplied as: A crystalline solid Storage: -20°C Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Remogliflozin A is supplied as a crystalline solid. A stock solution may be made by dissolving the remogliflozin A in the solvent of choice. Remogliflozin A is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide, which should be purged with an inert gas. The solubility of remogliflozin A in these solvents is approximately 30 mg/ml. Remogliflozin A is sparingly soluble in aqueous buffers. For maximum solubility in aqueous buffers, remogliflozin A should first be dissolved in ethanol and then diluted with the aqueous buffer of choice. Remogliflozin A has a solubility of approximately 0.5 mg/ml in a 1:1 solution of ethanol:PBS (pH 7.2) using this method. We do not recommend storing the aqueous solution for more than one day. Description Remogliflozin A is a potent inhibitor of sodium-glucose transporter 2 (SGLT2; Kis = 12.4 and 26 nM 1 for human and rat SGLT2, respectively). It is selective for SGLT2 over SGLT1 (Kis = 4,520 and 997 nM for human and rat SGLT1, respectively). Following administration of a prodrug, remogliflozin etabonate, that is rapidly converted to remogliflozin A in vivo, rat urinary glucose excretion increases and plasma glucose and insulin concentrations decrease. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Effect of Oral Hypoglycaemic Agents on Glucose Tolerance in Pancreatic Diabetes
Gut: first published as 10.1136/gut.13.4.285 on 1 April 1972. Downloaded from Gut, 1972, 13, 285-288 Effect of oral hypoglycaemic agents on glucose tolerance in pancreatic diabetes B. I. JOFFE, W. P. U. JACKSON, S. BANK, AND A. I. VINIK From the Department of Medicine, Witwatersrand University Medical School, Johannesburg, the Gastro- intestinal and Endocrine Research Units of Cape Town University Medical School, and the Chemical Pathology Department of Natal University, South Africa SUMMARY The short-term therapeutic effect of oral hypoglycaemic agents has been assessed in 12 patients with symptomatic diabetes secondary to chronic pancreatitis (pancreatic diabetes). In six patients who had moderate to severe carbohydrate intolerance, associated with severe insulino- paenia during arginine infusion, the potent sulphonylurea chlorpropamide produced no change in the fasting blood glucose level after two weeks of treatment. This contrasted with the significant reduction produced in a matched group of maturity-onset primary diabetics. The six patients with milder diabetes, and a greater (although still subnormal) insulin secretory capacity, showed an improvement in oral glucose tolerance during the first hour following glucose administration while on chlorpropamide. When the biguanide phenformin was substituted for chlorpropamide in five of these patients, a statistically insignificant improvement in glucose tolerance was observed during treatment. Applications of these findings to the practical management of pancreatic diabetes are briefly http://gut.bmj.com/ considered. Chronic pancreatitis is frequently complicated by and two women, ranging from 30 to 67 years of age. diabetes (pancreatic diabetes). Recent studies The diagnosis of pancreatitis was confirmed on the utilizing immunoassay procedures (Joffe, Bank, basis of a gross abnormality in at least two aspects of Jackson, Keller, O'Reilly, and Vinik, 1968; Anderson the pancreatic function test, namely, a low volume of on September 24, 2021 by guest. -

Remogliflozin Etabonate, a Selective Inhibitor of the Sodium-Glucose
Clinical Care/Education/Nutrition/Psychosocial Research BRIEF REPORT Remogliflozin Etabonate, a Selective Inhibitor of the Sodium-Glucose Transporter 2, Improves Serum Glucose Profiles in Type 1 Diabetes 1,2 3 SUNDER MUDALIAR, MD JUNE YE, PHD placebo (placebo), 2) mealtime insulin 1 3 DEBRA A. ARMSTRONG, BA, RN, CCRC ELIZABETH K. HUSSEY, PHARMD 2 3 injection + RE placebo (prandial insulin), ANNIE A. MAVIAN, MD DEREK J. NUNEZ, MD 3 3 1,2 ) placebo insulin injection + 50 mg RE (RE ROBIN O’CONNOR-SEMMES, PHD ROBERT R. HENRY, MD 3 3 50 mg), 4) placebo insulin injection + 150 PATRICIA K. MYDLOW, BS ROBERT L. DOBBINS, MD, PHD mg RE (RE 150 mg), and 5) placebo insulin injection + 500 mg RE (RE 500 mg). d fl Each individual received 75-g oral OBJECTIVES Remogli ozin etabonate (RE), an inhibitor of the sodium-glucose transporter glucose and identical meals during all 2, improves glucose profiles in type 2 diabetes. This study assessed safety, tolerability, pharma- cokinetics, and pharmacodynamics of RE in subjects with type 1 diabetes. treatment periods. Frequent samples were obtained for measurement of plasma RESEARCH DESIGN AND METHODSdTen subjects managed with continuous sub- glucose and insulin concentrations. Urine cutaneous insulin infusion were enrolled. In addition to basal insulin, subjects received five samples were collected for 24 h to assess randomized treatments: placebo, prandial insulin, 50 mg RE, 150 mg RE, and mg RE 500. creatinine clearance and glucose excre- d tion. Plasma samples were collected for RESULTS Adverse events and incidence of hypoglycemia with RE did not differ from placebo fl and prandial insulin groups. -

Diabetes-Related Biomarkers and Methods of Use Thereof
(19) TZZ _ _T (11) EP 2 541 254 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: G01N 21/76 (2006.01) G01N 21/64 (2006.01) 12.11.2014 Bulletin 2014/46 G01N 33/66 (2006.01) G01N 33/74 (2006.01) G01N 33/68 (2006.01) (21) Application number: 12175286.9 (22) Date of filing: 18.04.2008 (54) Diabetes-related biomarkers and methods of use thereof Diabetesassoziierte Biomarker und Verfahren zur deren Verwendung dafür Biomarqueurs associés aux diabètes et procédés d’utilisation correspondants (84) Designated Contracting States: • HANLEY ANTHONY J G ET AL: "Metabolic and AT BE BG CH CY CZ DE DK EE ES FI FR GB GR inflammation variable clusters and prediction of HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT type 2 diabetes: factor analysis using directly RO SE SI SK TR measured insulin sensitivity.", DIABETES JUL 2004, vol. 53, no. 7, July 2004 (2004-07), pages (30) Priority: 18.04.2007 US 788260 1773-1781, XP002486180, ISSN: 0012-1797 08.11.2007 US 2609 P • EDELMAN DAVID ET AL: "Utility of hemoglobin A1c in predicting diabetes risk", JOURNAL OF (43) Date of publication of application: GENERAL INTERNAL MEDICINE, vol. 19, no. 12, 02.01.2013 Bulletin 2013/01 December 2004 (2004-12), pages 1175-1180, XP002502995, ISSN: 0884-8734 (62) Document number(s) of the earlier application(s) in • INOUE ET AL: "The combination of fasting accordance with Art. 76 EPC: plasma glucose and glycosylated hemoglobin 08746276.8 / 2 147 315 predicts type 2 diabetes in Japanese workers", DIABETES RESEARCH AND CLINICAL (73) Proprietor: Health Diagnostic Laboratory, Inc. -

Dipeptidyl Peptidase-4 Inhibitors and Combinations
Dipeptidyl Peptidase-4 Inhibitors & Combinations Policy Number: C5169A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DATE 06/2016 10/30/2019 10/30/2020 J CODE TYPE OF CRITERIA LAST P&T APPROVAL/VERSION NA RxPA Q4 2019 20191030C5169-A PRODUCTS AFFECTED: KAZANO (alogliptin/metformin), KOMBIGLYZE XR (saxagliptin/metformin extended-release), NESINA (alogliptin), ONGLYZA (saxagliptin), OSENI (alogliptin/pioglitazone), JANUMET (sitagliptin/metformin), JANUMET XR (sitagliptin/metformin extended-release), JANUVIA (sitagliptin), JENTADUETO (linagliptin/metformin), JENTADUETO XR (linagliptin/metformin extended-release), KAZANO (alogliptin/metformin), KOMBIGLYZE XR (saxagliptin/metformin extended-release), NESINA (alogliptin), ONGLYZA (saxagliptin), OSENI (alogliptin/pioglitazone) TRADJENTA (linagliptin), JANUVIA (sitagliptin) DRUG CLASS: Dipeptidyl Peptidase-4 Inhibitor-(Biguanide Combinations), DPP-4 Inhibitor- Thiazolidinedione Combinations ROUTE OF ADMINISTRATION: Oral PLACE OF SERVICE: Retail Pharmacy AVAILABLE DOSAGE FORMS: Alogliptin Benzoate TABS 12.5MG,Alogliptin Benzoate TABS 25MG, Alogliptin Benzoate TABS 6.25MG, Alogliptin-Metformin HCl TABS 12.5-1000MG Alogliptin-Metformin HCl TABS 12.5-500MG, Janumet TABS 50-1000MG, Janumet TABS 50- 500MG, Janumet XR TB24 100-1000MG, Janumet XR TB24 50-1000MG, Janumet XR TB24 50- 500MG, Januvia TABS 100MG, Januvia TABS 25MG, Januvia TABS 50MG, Jentadueto TABS 2.5- 1000MG, Jentadueto TABS 2.5-500MG, Jentadueto TABS 2.5-500MG, Jentadueto TABS 2.5- 850MG, Jentadueto -

Englitazone Administration to Late Pregnant Rats Produces Delayed Body Growth and Insulin Resistance in Their Fetuses and Neonates
Biochem. J. (2005) 389, 913–918 (Printed in Great Britain) 913 Englitazone administration to late pregnant rats produces delayed body growth and insulin resistance in their fetuses and neonates Julio SEVILLANO, Inmaculada C. LOPEZ-P´ EREZ,´ Emilio HERRERA, Mar´ıa DEL PILAR RAMOS and Carlos BOCOS1 Facultad de Farmacia, Universidad San Pablo-CEU, Montepr´ıncipe, Ctra. Boadilla del Monte Km 5.300, E-28668 Boadilla del Monte (Madrid), Spain The level of maternal circulating triacylglycerols during late preg- pregnant rats was corroborated, since they showed higher plasma nancy has been correlated with the mass of newborns. PPARγ NEFA [non-esterified (‘free’) fatty acid] levels, ketonaemia and (peroxisome-proliferator-activated receptor γ ) ligands, such as liver LPL (lipoprotein lipase) activity and lower plasma IGF-I TZDs (thiazolidinediones), have been shown to reduce triacyl- (type 1 insulin-like growth factor) levels, in comparison with glycerolaemia and have also been implicated in the inhibition those from control mothers. Moreover, at the molecular level, an of tissue growth and the promotion of cell differentiation. increase in Akt phosphorylation was found in the liver of neonates Therefore TZDs might control cell proliferation during late fetal from EZ-treated mothers, which confirms that the insulin pathway development and, by extension, body mass of pups. To investigate was negatively affected. Thus the response of fetuses and neonates the response to EZ (englitazone), a TZD, on perinatal develop- to maternal antidiabetic drug treatment is the opposite of what ment, 0 or 50 mg of englitazone/kg of body mass was given as an would be expected, and can be justified by the scarce amount of oral dose to pregnant rats daily from day 16 of gestation until either adipose tissue impeding a normal response to PPARγ ligands and day 20 for the study of their fetuses, or until day 21 of gestation by hyperinsulinaemia as being responsible for a major insulin- for the study of neonates. -
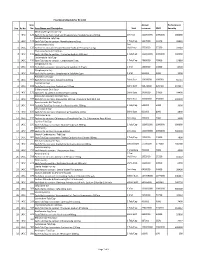
Sno. Rc No Item No. Item Name and Description Unit Annual Turnover
Final Drug Schedule for RC 145C Item Annual Performance Sno. Rc No No. Item Name and Description Unit turnover EMD Security Medroxy Progesterone Inj- 1 145C 126 Each ml to contain: Medroxy Progesterone Acetate Suspn.150mg. 1ml Vial 100000000 1000000 600000 Norethisterone Tab/Cap- 2 145C 128 Each Tab/Cap to contain: Norethisterone 5mg. 1 Tab/Cap 5637000 56370 33822 Dexamethasone Inj- 2ml 3 145C 133 Each ml to contain: Dexamethasone Sodium Phosphate 4 mg. Vial/Amp 5735000 57350 34410 Thyroxine Sodium Tab/Cap- 4 145C 135 Each Tab/Cap to contain: Thyroxine Sodium 100 mcg 1 Tab/Cap 100000000 1000000 600000 Carbimazole Tab/Cap- 5 145C 136 Each Tab/Cap to contain: Carbimazole 5 mg. 1 Tab/Cap 2980000 29800 17880 Streptomycin Inj- 6 145C 151 Each Vial to contain: Streptomycin Sulphate 0.75gm 1 Vial 1400000 14000 8400 Streptomycin Inj- 7 145C 152 Each Vial to contain: Streptomycin Sulphate 1gm 1 Vial 500000 5000 3000 Ampicillin Dry Syp- 8 145C 161 Each 5ml to contain: Ampicillin 125mg 30ml Bott 10959000 109590 65754 Cephalexin Syp- 9 145C 168 Each 5ml to contain: Cephalexin 125mg 30ml Bott 32874000 328740 197244 Erthyromycin Oral Susp- 10 145C 172 Each 5ml to contain: Erythromycin 125mg 30ml Bott 2400000 24000 14400 Amoxy & Clavulanic Acid Dry Syp- 11 145C 183 Each 5ml to contain: Amoxycillin 200 mg, Clavulanic Acid 28.5 mg 30ml Bott 35000000 350000 210000 Pyrazinamide Kid Tab/Cap- 12 145C 191 Each Kid Tab/Cap to contain: Pyrazinamide 300mg 1 Tab/Cap 500000 5000 3000 Chloroquine Syp- 13 145C 196 Each 5ml to contain: Chloroquine Phosphate 50mg. -

Polyhexamethylene Biguanide Hydrochloride) As Used in Cosmetics
Safety Assessment of Polyaminopropyl Biguanide (polyhexamethylene biguanide hydrochloride) as Used in Cosmetics Status: Tentative Report for Public Comment Release Date: September 26, 2017 Panel Date: December 4-5, 2017 All interested persons are provided 60 days from the above date to comment on this safety assessment and to identify additional published data that should be included or provide unpublished data which can be made public and included. Information may be submitted without identifying the source or the trade name of the cosmetic product containing the ingredient. All unpublished data submitted to CIR will be discussed in open meetings, will be available at the CIR office for review by any interested party and may be cited in a peer-reviewed scientific journal. Please submit data, comments, or requests to the CIR Executive Director, Dr. Bart Heldreth. The 2017 Cosmetic Ingredient Review Expert Panel members are: Chair, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Executive Director is Bart Heldreth, Ph.D. This report was prepared by Wilbur Johnson, Jr., M.S., Senior Scientific Analyst and Ivan Boyer, Ph.D., former Senior Toxicologist. © Cosmetic Ingredient Review 1620 L STREET, NW, SUITE 1200 ◊ WASHINGTON, DC 20036-4702 ◊ PH 202.331.0651 ◊ FAX 202.331.0088 ◊ [email protected] ABSTRACT: The Cosmetic Ingredient Review (CIR) Expert Panel (Panel) reviewed the safety of Polyaminopropyl Biguanide (polyhexamethylene biguanide hydrochloride), which functions as a preservative in cosmetic products.