Unix Intro Exercises
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bash Scripts for Avpres Verify Manifest(1)
verify_manifest(1) Bash Scripts for AVpres verify_manifest(1) NAME verify_manifest - Verify a checksum manifest of a folder or file SYNOPSIS verify_manifest -i input_path [-m manifest_file] verify_manifest -h | -x DESCRIPTION Bash AVpres is a collection of Bash scripts for audio-visual preservation. One of these small programs is verify_manifest.Itcreates a checksum manifest of a folder or file. Bash version 3.2 is required, but we strongly advise to use the current version 5.1. OPTIONS BASIC OPTIONS -i input_path,--input=input_path path to an input folder or file -m manifest_file,--manifest=manifest_file path to the manifest file If this parameter is not passed, then the script uses for a folder: <input_path>_<algorithm>.txt and for a file: <input_path>_<extension>_<algorithm>.txt ADVA NCED OPTIONS The arguments of the advanced options can be overwritten by the user.Please remember that anystring containing spaces must be quoted, or its spaces must be escaped. --algorithm=(xxh32|xxh64|xxh128|md5|sha1|sha256|sha512|crc32) We advise to use a faster non-cryptographic hash functions, because we consider that, for archival purposes, there is no necessity to apply a more complexunkeyed cryptographic hash function. The algorithm name can be passed in upper or lower case letters. The default algorithm is xxHash 128: --algorithm=xxh128 Note that until end of 2020 the default algorithm was MD5, which has the same checksum size than the xxHash 128 algorithm. Therefore, if you verity files with an MD5 checksum, then you may pass the option --algorithm=md5 in order to speed-up the verification. Also xxHash 32 and CRC-32 have the same checksum size. -

Download Instructions—Portal
Download instructions These instructions are recommended to download big files. How to download and verify files from downloads.gvsig.org • H ow to download files • G NU/Linux Systems • MacO S X Systems • Windows Systems • H ow to validate the downloaded files How to download files The files distributed on this site can be downloaded using different access protocols, the ones currently available are FTP, HTTP and RSYNC. The base URL of the site for the different protocols is: • ftp://gvsig.org/ • http://downloads.gvsig.org/ • r sync://gvsig.org/downloads/ To download files using the first two protocols is recommended to use client programs able to resume partial downloads, as it is usual to have transfer interruptions when downloading big files like DVD images. There are multiple free (and multi platform) programs to download files using different protocols (in our case we are interested in FTP and HTTP), from them we can highlight curl (http://curl.haxx.se/) and wget (http://www.gnu.org/software/wget/) from the command line ones and Free Download Manager from the GUI ones (this one is only for Windows systems). The curl program is included in MacOS X and is available for almost all GNU/Linux distributions. It can be downloaded in source code or in binary form for different operating systems from the project web site. The wget program is also included in almost all GNU/Linux distributions and its source code or binaries of the program for different systems can be downloaded from this page. Next we will explain how to download files from the most usual operating systems using the programs referenced earlier: • G NU/Linux Systems • MacO S X Systems • Windows Systems The use of rsync (available from the URL http://samba.org/rsync/) it is left as an exercise for the reader, we will only said that it is advised to use the --partial option to avoid problems when there transfers are interrupted. -
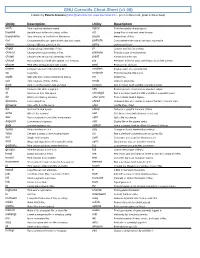
GNU Coreutils Cheat Sheet (V1.00) Created by Peteris Krumins ([email protected], -- Good Coders Code, Great Coders Reuse)
GNU Coreutils Cheat Sheet (v1.00) Created by Peteris Krumins ([email protected], www.catonmat.net -- good coders code, great coders reuse) Utility Description Utility Description arch Print machine hardware name nproc Print the number of processors base64 Base64 encode/decode strings or files od Dump files in octal and other formats basename Strip directory and suffix from file names paste Merge lines of files cat Concatenate files and print on the standard output pathchk Check whether file names are valid or portable chcon Change SELinux context of file pinky Lightweight finger chgrp Change group ownership of files pr Convert text files for printing chmod Change permission modes of files printenv Print all or part of environment chown Change user and group ownership of files printf Format and print data chroot Run command or shell with special root directory ptx Permuted index for GNU, with keywords in their context cksum Print CRC checksum and byte counts pwd Print current directory comm Compare two sorted files line by line readlink Display value of a symbolic link cp Copy files realpath Print the resolved file name csplit Split a file into context-determined pieces rm Delete files cut Remove parts of lines of files rmdir Remove directories date Print or set the system date and time runcon Run command with specified security context dd Convert a file while copying it seq Print sequence of numbers to standard output df Summarize free disk space setuidgid Run a command with the UID and GID of a specified user dir Briefly list directory -

Addresstranslation Adminguide
Address Translation Administrator Guide This guide is for administrators of MessageLabs Email Services. This guide is for administrators of MessageLabs Email Services. The Address Translation service is a MessageLabs Email Services feature that enables external internet-routable email addresses to be converted into internally-routable addresses, and vice versa. Document version 1.0 2006-12-07 Table of Contents 1 About the guide 3 1.1 Audience and scope 3 1.2 Versions of this guide 3 1.3 Conventions 3 2 Introduction to Address Translation 4 3 Configuring Address Translation 5 3.1 Formatting configuration data 5 3.2 Providing configuration data in CSV files 5 4 Uploading addrtrans.csv 6 4.1 Generating sha1sums (Linux) 6 4.2 Generating sha1sums (Windows) 6 4.3 Uploading CSV files to a Linux server 7 4.4 Uploading CSV files to a Windows server 7 5 Maintaining Address Translation data 8 2 1 About the guide 1.1 Audience and scope This guide is for administrators of MessageLabs Email Services. The Address Translation service is a MessageLabs Email Services feature that enables external internet-routable email addresses to be converted into internally-routable addresses, and vice versa. 1.2 Versions of this guide This guide is available in two page sizes: Letter (279 mm x 215.9 mm) and A4 (297 mm x 210 mm). The version is identified at the end of the file name as _Ltr or _A4. The content is identical in the two versions. Use whichever suits your printing requirements. 1.3 Conventions In this guide, the following conventions are used: Formatting Denotes Bold Button, tab or field Bold Italic Window title or description Note: A note containing extra information that may be useful Text to type in Text to type in Output from a computer Output from a computer Link A link to a website Screenshots normally display an Internet Explorer window. -

Learn Programming
Learn Programming Antti Salonen Aug 04, 2018 Learn Programming, first edition Antti Salonen This work is licensed under the Creative Commons Attribution-ShareAlike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-sa/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA. The code within this book is licensed under the MIT license. Copyright 2018, Antti Salonen. 2 CONTENTS: 1 The beginning 7 1.1 Introduction ......................... 8 1.1.1 Why this book? .................... 8 1.1.2 What is software? .................. 15 1.1.3 How does a computer work? ............ 19 1.1.4 OK, but seriously, how does a computer work? ... 26 1.1.5 The basics of programming ............. 30 1.1.6 Setting up the C toolchain .............. 39 1.1.7 The basics of programming in C .......... 43 1.1.8 Learning to learn .................. 53 1.2 Basics of programming in Python and C .......... 56 1.2.1 Quadratic formula in C ............... 56 1.2.2 Lots of quadratic equations ............. 60 1.2.3 Quadratic formula in Python ............ 65 1.2.4 Generating input data using Python ........ 71 1.3 Unix shell ........................... 72 1.3.1 Basic Unix shell usage ................ 72 1.3.2 Unix shell scripting ................. 82 1.3.3 Regular expressions ................. 89 1.4 Using libraries in Python ................... 93 1.4.1 Creating a simple web page ............. 93 1.4.2 Making our web page work ............. 97 2 Stage 1 103 2.1 Further Unix tools ...................... 104 2.1.1 Version control .................. -

Puremessage for Unix Help Contents Getting Started
PureMessage for Unix help Contents Getting Started......................................................................................................................................... 1 Welcome to PureMessage for Unix.............................................................................................. 1 Deployment Strategies.................................................................................................................. 6 Installing PureMessage............................................................................................................... 18 Upgrading PureMessage.............................................................................................................51 Quick Reference Guide...............................................................................................................65 Contacting Sophos...................................................................................................................... 75 Managing PureMessage........................................................................................................................ 77 Dashboard Tab............................................................................................................................77 Policy Tab....................................................................................................................................79 Quarantine Tab..........................................................................................................................130 -

Classic Shell Scripting
Classic Shell Scripting Arnold Robbins and Nelson H. F. Beebe Beijing • Cambridge • Farnham • Köln • Sebastopol • Tokyo Classic Shell Scripting by Arnold Robbins and Nelson H. F. Beebe Copyright © 2005 O’Reilly Media, Inc. All rights reserved. Printed in the United States of America. Published by O’Reilly Media, Inc., 1005 Gravenstein Highway North, Sebastopol, CA 95472. O’Reilly books may be purchased for educational, business, or sales promotional use. Online editions are also available for most titles (safari.oreilly.com). For more information,contact our corporate/insti- tutional sales department: (800) 998-9938 or [email protected]. Editors: Tatiana Apandi Allison Randal Production Editor: Adam Witwer Cover Designer: Emma Colby Interior Designer: David Futato Printing History: May 2005: First Edition. Nutshell Handbook, the Nutshell Handbook logo, and the O’Reilly logo are registered trademarks of O’Reilly Media,Inc. Classic Shell Scripting,the image of a African tent tortoise,and related trade dress are trademarks of O’Reilly Media, Inc. Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. Where those designations appear in this book, and O’Reilly Media, Inc. was aware of a trademark claim, the designations have been printed in caps or initial caps. While every precaution has been taken in the preparation of this book, the publisher and authors assume no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. ISBN: 978-0-596-00595-5 [LSI] [2011-03-11] Table of Contents Foreword . ix Preface . xi 1. Background . 1 1.1 Unix History 1 1.2 Software Tools Principles 4 1.3 Summary 6 2. -

Gnu Coreutils Core GNU Utilities for Version 5.93, 2 November 2005
gnu Coreutils Core GNU utilities for version 5.93, 2 November 2005 David MacKenzie et al. This manual documents version 5.93 of the gnu core utilities, including the standard pro- grams for text and file manipulation. Copyright c 1994, 1995, 1996, 2000, 2001, 2002, 2003, 2004, 2005 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.1 or any later version published by the Free Software Foundation; with no Invariant Sections, with no Front-Cover Texts, and with no Back-Cover Texts. A copy of the license is included in the section entitled “GNU Free Documentation License”. Chapter 1: Introduction 1 1 Introduction This manual is a work in progress: many sections make no attempt to explain basic concepts in a way suitable for novices. Thus, if you are interested, please get involved in improving this manual. The entire gnu community will benefit. The gnu utilities documented here are mostly compatible with the POSIX standard. Please report bugs to [email protected]. Remember to include the version number, machine architecture, input files, and any other information needed to reproduce the bug: your input, what you expected, what you got, and why it is wrong. Diffs are welcome, but please include a description of the problem as well, since this is sometimes difficult to infer. See section “Bugs” in Using and Porting GNU CC. This manual was originally derived from the Unix man pages in the distributions, which were written by David MacKenzie and updated by Jim Meyering. -

Software Release Notes for 5.0
Software Release Notes for 5.0 Jeffery A. Triggs, Sho Nakagama, & Isaiah Beard August 17, 2009 1 Introduction This document supersedes relevant sections of the one entitled “Software Release Procedures for 1.1” dated February 8, 2006. It reflects our our migration to a new platform, including Fedora 3.0, PHP 5, and MySQL 5. The following sections describe: 1) the base software platform, which should already be in place before any release of the project software; 2) the different projects to be released along with their functions. 2 Basic Software Platform Requirements This section deals with the underlying software platforms necessary for the 5.0 release. The software is developed on Linux at the SCC and released on Solaris at Library Systems. The basic software packages are compiled and loaded separately on each of these platforms, and should remain stable throughout the 5.x release cycle. 2.1 amberfish 1.6.4 2.1.1 xerces 2.7 2.2 Apache Server version: Apache/2.0.59 Server compiled with.... -D APACHE_MPM_DIR="server/mpm/prefork" Software Release Notes for 5.0 2 -D APR_HAS_SENDFILE -D APR_HAS_MMAP -D APR_HAVE_IPV6 (IPv4-mapped addresses enabled) -D APR_USE_SYSVSEM_SERIALIZE -D APR_USE_PTHREAD_SERIALIZE -D SINGLE_LISTEN_UNSERIALIZED_ACCEPT -D APR_HAS_OTHER_CHILD -D AP_HAVE_RELIABLE_PIPED_LOGS -D HTTPD_ROOT="/usr/local/apache2" -D SUEXEC_BIN="/usr/local/apache2/bin/suexec" -D DEFAULT_PIDLOG="logs/httpd.pid" -D DEFAULT_SCOREBOARD="logs/apache_runtime_status" -D DEFAULT_LOCKFILE="logs/accept.lock" -D DEFAULT_ERRORLOG="logs/error_log" -D AP_TYPES_CONFIG_FILE="conf/mime.types" -D SERVER_CONFIG_FILE="conf/httpd.conf" ./configure --enable-ssl --enable-so --with-mpm=prefork --enable-mods-shared=most --with-openssl=/usr 2.3 CNRI Handles The CNRI handle client is run typically on ingest. -

Pash: Light-Touch Data-Parallel Shell Processing
PaSh: Light-touch Data-Parallel Shell Processing Nikos Vasilakis∗ Konstantinos Kallas∗ Konstantinos Mamouras MIT University of Pennsylvania Rice University [email protected] [email protected] [email protected] Achilles Benetopoulos Lazar Cvetković Unaffiliated University of Belgrade [email protected] [email protected] Abstract Parallelizability Parallelizing Runtime Classes §3 Transformations §4.3 Primitives §5 Dataflow This paper presents PaSh, a system for parallelizing POSIX POSIX, GNU §3.1 Regions shell scripts. Given a script, PaSh converts it to a dataflow Annotations §3.2 § 4.1 DFG § 4.4 graph, performs a series of semantics-preserving program §4.2 transformations that expose parallelism, and then converts Seq. Script Par. Script the dataflow graph back into a script—one that adds POSIX constructs to explicitly guide parallelism coupled with PaSh- Fig. 1. PaSh overview. PaSh identifies dataflow regions (§4.1), converts provided Unix-aware runtime primitives for addressing per- them to dataflow graphs (§4.2), applies transformations (§4.3) based onthe parallelizability properties of the commands in these regions (§3.1, §3.2), formance- and correctness-related issues. A lightweight an- and emits a parallel script (§4.4) that uses custom primitives (§5). notation language allows command developers to express key parallelizability properties about their commands. An accompanying parallelizability study of POSIX and GNU • Command developers, responsible for implementing indi- commands—two large and commonly used groups—guides vidual commands such as sort, uniq, and jq. These de- the annotation language and optimized aggregator library velopers usually work in a single programming language, that PaSh uses. PaSh’s extensive evaluation over 44 unmod- leveraging its abstractions to provide parallelism when- ified Unix scripts shows significant speedups (0.89–61.1×, ever possible. -

Linux Shell Scripting Cookbook Second Edition
Linux Shell Scripting Cookbook Second Edition Over 110 practical recipes to solve real-world shell problems, guaranteed to make you wonder how you ever lived without them Shantanu Tushar Sarath Lakshman BIRMINGHAM - MUMBAI Linux Shell Scripting Cookbook Second Edition Copyright © 2013 Packt Publishing All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, without the prior written permission of the publisher, except in the case of brief quotations embedded in critical articles or reviews. Every effort has been made in the preparation of this book to ensure the accuracy of the information presented. However, the information contained in this book is sold without warranty, either express or implied. Neither the authors, nor Packt Publishing, and its dealers and distributors will be held liable for any damages caused or alleged to be caused directly or indirectly by this book. Packt Publishing has endeavored to provide trademark information about all of the companies and products mentioned in this book by the appropriate use of capitals. However, Packt Publishing cannot guarantee the accuracy of this information. First published: January 2011 Second edition: May 2013 Production Reference: 1140513 Published by Packt Publishing Ltd. Livery Place 35 Livery Street Birmingham B3 2PB, UK. ISBN 978-1-78216-274-2 www.packtpub.com Cover Image by Parag Kadam ([email protected]) Credits Authors Project Coordinator Shantanu Tushar Shiksha Chaturvedi Sarath Lakshman Proofreader Reviewers Linda Morris Rajeshwari K. John C. Kennedy Indexer Hemangini Bari Anil Kumar Sudhendu Kumar Production Coordinator Aravind SV Shantanu Zagade Acquisition Editor Cover Work Kartikey Pandey Shantanu Zagade Lead Technical Editor Ankita Shashi Technical Editors Jalasha D'costa Amit Ramadas Lubna Shaikh About the Authors Shantanu Tushar is an advanced GNU/Linux user since his college days. -

WBAN) for Healthcare
International Journal of Research in Engineering, Science and Management 614 Volume-2, Issue-3, March-2019 www.ijresm.com | ISSN (Online): 2581-5792 QOS based Wireless Body Area Network (WBAN) for Healthcare P. Kavitha1, R. Jayasanthi2, S. Meenakshi Ishwaryar3 1Assistant Professor, Dept. of CSE, P. S. R. Rengasamy College of Engineering for Women, Sivakasi, India 2,3UG Student, Dept. of CSE, P. S. R. Rengasamy College of Engineering for Women, Sivakasi, India Abstract: In many military network scenarios, connections of Delays can be typically on the order of minutes or wire- less devices carried by soldiers may be temporarily hours, but could potentially be days depending on the disconnected by jamming, environmental factors, and mobility, exact scenario. especially when they operate in hostile environments. In a hospital environment, the total number of Wireless Body Area Network C. Objectives (WBAN) equipped patients requesting ubiquitous healthcare services in an area increases significantly. Therefore, increased Asynchronously interconnecting different networks traffic load and group-based mobility of WBANs degrades the Network of regional networks Network of regional performance of each WBAN significantly, concerning service networks delay and network throughput. In addition, the mobility of Each network can have WBANs affects connectivity between a WBAN and an Access Arbitrary underlying technologies Point (AP) dynamically, which affects the variation in link quality significantly. To address the connectivity problem and provide Different administrative controls Quality of Services (QoS) in the network, we propose a dynamic No accessible infrastructure connectivity establishment and cooperative scheduling scheme, which minimizes the packet delivery delay and maximizes the 2. Existing system network throughput.