Oxidation Catalytic System and Oxidation Process Using Same
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

First Principles Prediction of Thermodynamic Properties
2 First Principles Prediction of Thermodynamic Properties Hélio F. Dos Santos and Wagner B. De Almeida NEQC: Núcleo de Estudos em Química Computacional, Departamento de Química, ICE Universidade Federal de Juiz de Fora (UFJF), Campus Universitário Martelos, Juiz de Fora LQC-MM: Laboratório de Química Computacional e Modelagem Molecular Departamento de Química, ICEx, Universidade Federal de Minas Gerais (UFMG) Campus Universitário, Pampulha, Belo Horizonte Brazil 1. Introduction The determination of the molecular structure is undoubtedly an important issue in chemistry. The knowledge of the tridimensional structure allows the understanding and prediction of the chemical-physics properties and the potential applications of the resulting material. Nevertheless, even for a pure substance, the structure and measured properties reflect the behavior of many distinct geometries (conformers) averaged by the Boltzmann distribution. In general, for flexible molecules, several conformers can be found and the analysis of the physical and chemical properties of these isomers is known as conformational analysis (Eliel, 1965). In most of the cases, the conformational processes are associated with small rotational barriers around single bonds, and this fact often leads to mixtures, in which many conformations may exist in equilibrium (Franklin & Feltkamp, 1965). Therefore, the determination of temperature-dependent conformational population is very much welcomed in conformational analysis studies carried out by both experimentalists and theoreticians. -

Comparing Models for Measuring Ring Strain of Common Cycloalkanes
The Corinthian Volume 6 Article 4 2004 Comparing Models for Measuring Ring Strain of Common Cycloalkanes Brad A. Hobbs Georgia College Follow this and additional works at: https://kb.gcsu.edu/thecorinthian Part of the Chemistry Commons Recommended Citation Hobbs, Brad A. (2004) "Comparing Models for Measuring Ring Strain of Common Cycloalkanes," The Corinthian: Vol. 6 , Article 4. Available at: https://kb.gcsu.edu/thecorinthian/vol6/iss1/4 This Article is brought to you for free and open access by the Undergraduate Research at Knowledge Box. It has been accepted for inclusion in The Corinthian by an authorized editor of Knowledge Box. Campring Models for Measuring Ring Strain of Common Cycloalkanes Comparing Models for Measuring R..ing Strain of Common Cycloalkanes Brad A. Hobbs Dr. Kenneth C. McGill Chemistry Major Faculty Sponsor Introduction The number of carbon atoms bonded in the ring of a cycloalkane has a large effect on its energy. A molecule's energy has a vast impact on its stability. Determining the most stable form of a molecule is a usefol technique in the world of chemistry. One of the major factors that influ ence the energy (stability) of cycloalkanes is the molecule's ring strain. Ring strain is normally viewed as being directly proportional to the insta bility of a molecule. It is defined as a type of potential energy within the cyclic molecule, and is determined by the level of "strain" between the bonds of cycloalkanes. For example, propane has tl1e highest ring strain of all cycloalkanes. Each of propane's carbon atoms is sp3-hybridized. -

Text Related to Segment 5.02 ©2002 Claude E. Wintner from the Previous Segment We Have the Value of the Heat of Combustion Of
Text Related to Segment 5.02 ©2002 Claude E. Wintner From the previous segment we have the value of the heat of combustion of an "unstrained" methylene unit as -157.4 kcal/mole. On this basis one would predict that combustion of "unstrained" cyclopropane should liberate 3 X 157.4 = 472.2 kcal/mole; however, when cyclopropane is burned, a value of 499.8 kcal/mole is measured for the actual heat of combustion. The difference of 27.6 kcal/mole is interpreted as representing the higher internal energy ("strain energy") of real cyclopropane as compared to a postulated strain-free "model." ("Model" in this usage speaks of a proposal, or postulate.) These facts are graphed conveniently on an energy diagram as follows: "Strain Energy" represents the error units: kcal/mole not to scale in the "strain-free model" real cyclopropane + 4.5 O2 27.6 "strain-free model" of cyclopropane 499.8 observed heat of combustion + 4.5 O2 472.2 3CO2 + 3H2O heat of combustion predicted for "strain-free model" Note that this estimate of the strain energy in cyclopropane amounts to 9.2 kcal/mole of strain per methylene group (dividing 27.6 by 3, because of the three carbon atoms). Comparable values in kcal/mole for other small and medium rings are given in the following table. As one might expect, in general the strain decreases as the ring is enlarged. units: kcal/mole n Total Strain Strain per CH2 3 27.6 9.2 (CH2)n 4 26.3 6.6 5 6.2 1.2 6 0.1 0.0 ! 7 6.2 0.9 8 9.7 1.2 9 12.6 1.4 10 12.4 1.2 12 4.1 0.3 15 1.9 0.1 Without entering into a discussion of the relevant bonding concepts here, and instead relying on geometry alone, interpretation of the source of the strain energy in cyclopropane and cyclobutane is to some extent self-evident. -

An Indicator of Triplet State Baird-Aromaticity
inorganics Article The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub 1,2, Kjell Jorner 1,2 ID and Henrik Ottosson 1,2,* 1 Department of Chemistry—BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden; [email protected] (R.A.); [email protected] (K.J.) 2 Department of Chemistry-Ångström Laboratory Uppsala University, Box 523, SE-751 20 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-18-4717476 Received: 23 October 2017; Accepted: 11 December 2017; Published: 15 December 2017 Abstract: Baird’s rule tells that the electron counts for aromaticity and antiaromaticity in the first ππ* triplet and singlet excited states (T1 and S1) are opposite to those in the ground state (S0). Our hypothesis is that a silacyclobutene (SCB) ring fused with a [4n]annulene will remain closed in the T1 state so as to retain T1 aromaticity of the annulene while it will ring-open when fused to a [4n + 2]annulene in order to alleviate T1 antiaromaticity. This feature should allow the SCB ring to function as an indicator for triplet state aromaticity. Quantum chemical calculations of energy and (anti)aromaticity changes along the reaction paths in the T1 state support our hypothesis. The SCB ring should indicate T1 aromaticity of [4n]annulenes by being photoinert except when fused to cyclobutadiene, where it ring-opens due to ring-strain relief. Keywords: Baird’s rule; computational chemistry; excited state aromaticity; Photostability 1. Introduction Baird showed in 1972 that the rules for aromaticity and antiaromaticity of annulenes are reversed in the lowest ππ* triplet state (T1) when compared to Hückel’s rule for the electronic ground state (S0)[1–3]. -

The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”
chemengineering Article Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy” Robert J. Meier Pro-Deo Consultant, 52525 Heinsberg, North-Rhine Westphalia, Germany; [email protected] Abstract: Group contribution (GC) methods to predict thermochemical properties are of eminent importance to process design. Compared to previous works, we present an improved group contri- bution parametrization for the heat of formation of organic molecules exhibiting chemical accuracy, i.e., a maximum 1 kcal/mol (4.2 kJ/mol) difference between the experiment and model, while, at the same time, minimizing the number of parameters. The latter is extremely important as too many parameters lead to overfitting and, therewith, to more or less serious incorrect predictions for molecules that were not within the data set used for parametrization. Moreover, it was found to be important to explicitly account for common chemical knowledge, e.g., geminal effects or ring strain. The group-related parameters were determined step-wise: first, alkanes only, and then only one additional group in the next class of molecules. This ensures unique and optimal parameter values for each chemical group. All data will be made available, enabling other researchers to extend the set to other classes of molecules. Keywords: enthalpy of formation; thermodynamics; molecular modeling; group contribution method; quantum mechanical method; chemical accuracy; process design Citation: Meier, R.J. Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”. 1. Introduction ChemEngineering 2021, 5, 24. To understand chemical reactivity and/or chemical equilibria, knowledge of thermo- o https://doi.org/10.3390/ dynamic properties such as gas-phase standard enthalpy of formation DfH gas is a necessity. -

Cycloalkanes, Cycloalkenes, and Cycloalkynes
CYCLOALKANES, CYCLOALKENES, AND CYCLOALKYNES any important hydrocarbons, known as cycloalkanes, contain rings of carbon atoms linked together by single bonds. The simple cycloalkanes of formula (CH,), make up a particularly important homologous series in which the chemical properties change in a much more dramatic way with increasing n than do those of the acyclic hydrocarbons CH,(CH,),,-,H. The cyclo- alkanes with small rings (n = 3-6) are of special interest in exhibiting chemical properties intermediate between those of alkanes and alkenes. In this chapter we will show how this behavior can be explained in terms of angle strain and steric hindrance, concepts that have been introduced previously and will be used with increasing frequency as we proceed further. We also discuss the conformations of cycloalkanes, especially cyclo- hexane, in detail because of their importance to the chemistry of many kinds of naturally occurring organic compounds. Some attention also will be paid to polycyclic compounds, substances with more than one ring, and to cyclo- alkenes and cycloalkynes. 12-1 NOMENCLATURE AND PHYSICAL PROPERTIES OF CYCLOALKANES The IUPAC system for naming cycloalkanes and cycloalkenes was presented in some detail in Sections 3-2 and 3-3, and you may wish to review that ma- terial before proceeding further. Additional procedures are required for naming 446 12 Cycloalkanes, Cycloalkenes, and Cycloalkynes Table 12-1 Physical Properties of Alkanes and Cycloalkanes Density, Compounds Bp, "C Mp, "C diO,g ml-' propane cyclopropane butane cyclobutane pentane cyclopentane hexane cyclohexane heptane cycloheptane octane cyclooctane nonane cyclononane "At -40". bUnder pressure. polycyclic compounds, which have rings with common carbons, and these will be discussed later in this chapter. -

Alkenes and Alkynes
02/21/2019 CHAPTER FOUR Alkenes and Alkynes H N O I Cl C O C O Cl F3C C Cl C Cl Efavirenz Haloprogin (antiviral, AIDS therapeutic) (antifungal, antiseptic) Chapter 4 Table of Content * Unsaturated Hydrocarbons * Introduction and hybridization * Alkenes and Alkynes * Benzene and Phenyl groups * Structure of Alkenes, cis‐trans Isomerism * Nomenclature of Alkenes and Alkynes * Configuration cis/trans, and cis/trans Isomerism * Configuration E/Z * Physical Properties of Hydrocarbons * Acid‐Base Reactions of Hydrocarbons * pka and Hybridizations 1 02/21/2019 Unsaturated Hydrocarbons • Unsaturated Hydrocarbon: A hydrocarbon that contains one or more carbon‐carbon double or triple bonds or benzene‐like rings. – Alkene: contains a carbon‐carbon double bond and has the general formula CnH2n. – Alkyne: contains a carbon‐carbon triple bond and has the general formula CnH2n‐2. Introduction Alkenes ● Hydrocarbons containing C=C ● Old name: olefins • Steroids • Hormones • Biochemical regulators 2 02/21/2019 • Alkynes – Hydrocarbons containing C≡C – Common name: acetylenes Unsaturated Hydrocarbons • Arene: benzene and its derivatives (Ch 9) 3 02/21/2019 Benzene and Phenyl Groups • We do not study benzene and its derivatives until Chapter 9. – However, we show structural formulas of compounds containing a phenyl group before that time. – The phenyl group is not reactive under any of the conditions we describe in chapters 5‐8. Structure of Alkenes • The two carbon atoms of a double bond and the four atoms bonded to them lie in a plane, with bond angles of approximately 120°. 4 02/21/2019 Structure of Alkenes • Figure 4.1 According to the orbital overlap model, a double bond consists of one bond formed by overlap of sp2 hybrid orbitals and one bond formed by overlap of parallel 2p orbitals. -

Studies on Photochemical Reactions of Simple Alkenes
Title STUDIES ON PHOTOCHEMICAL REACTIONS OF SIMPLE ALKENES Author(s) 井上, 佳久 Citation Issue Date Text Version ETD URL http://hdl.handle.net/11094/1547 DOI rights Note Osaka University Knowledge Archive : OUKA https://ir.library.osaka-u.ac.jp/ Osaka University STUDIESON PHOTOCHEMICAL REACTIONS CF SIMPLE ALKENES YOSHIHISA INOUE OSAKA UNIVERSITY OSAKA, JAPAN JANUARY, 1977 ii Preface The work of thi's•theSis 'was performed under the guiqance of Professor Hiroshi Sakurai at the ZnstituteofScientific and lndustrial Research, Osaka University. I arn deeply grateÅíul to Professor Hiroshi Sakurai for his appro- priate guidance and continuOus encouragernent throughout this work since 1972. : am aiso grateful to Professor Yoshinobu Odaira-who initiated me into the organic photochemistry in 1971-1972. : arn indebted to Assistant Professor Setsuo Takamuku for his invaluable discussions throughout the work. Many thanks are given to Drs. Yoshiki Okarrtoto, Chyongjin Pac, Susumu Toki, and Yasuo Shige- mitsu for their helpful suggestions through the stimulating discussions with me. T would like to express my gratitude to Mr. Kazuyoshi Mori- tsugut Mr. Masahiro Kadohira, and Ms. Yoko Kunitomi for their collabo- rations in the course of experiment. Finally I wish to thank all the members of Sakurai Laboratory for their warm friendship. t- ..<2IZI Yoshihisa Inoue Suita, Osaka January, Z977 iii List of Papers The contents of this thesis are composed of the following papers. 1) Mercury Photosensitized Rbaction of Cycloheptene. Mechanism of Norcarane Formation Y. Tnoue, M. Kadohira, S. Takasnuku, and H. Sakurait Tetrahedron Lett., 459(1974). 2) Vapor-phase Photolysis of cis-- and trans-4i5-Diraethylcyclohexenes Y. rnoue, S. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -

Stereospecific Fragmentations in the Mass Spectra of Stereoisomeric Isoindoloquinazolines
Stereospecific Fragmentations in the Mass Spectra of Stereoisomeric Isoindoloquinazolines Vladimir V. Ovcharenko and Kalevi Pihlaja Structural Chemistry Group, Department of Chemistry, University of Turku, Turku, Finland Ge´za Sta´jer Institute of Pharmaceutical Chemistry, University of Szeged, Szeged, Hungary The mass spectrometric behavior of stereo- and regioisomeric, partially saturated isoindolo- quinazolines was studied by positive-ion electron ionization (EI) and fast-atom bombardment (FAB/LSIMS) mass spectrometry combined with collision-induced dissociation (CID). A highly stereospecific retro-Diels-Alder process was observed in the cyclohexene-fused isomers under the EI conditions, and a corresponding (although less specific) fragmentation was observed in their FAB spectra. In the absence of RDA fragmentations, regio- and stereoisomers of the cyclohexane-fused heterocycles could be distinguished based on their FAB/CID spectra. (J Am Soc Mass Spectrom 2003, 14, 1049–1056) © 2003 American Society for Mass Spectrometry arious condensed-skeleton heterocycles contain- behavior of these compounds under the EI and FAB ing up to three rings can be prepared starting conditions is reported in the present study. Vfrom stereoisomeric aminobicycloalkanecar- The choice of experimental methods was dictated by boxylic acids by applying a retro-Diels-Alder (RDA) the following considerations. Mass-spectrometric RDA decomposition as a part of the synthetic strategy. For fragmentations often proceed in a regio- or stereospe- instance, the reaction of 3-aminobicyclo[2.2.1]hept-5- cific manner [4, 5]. The structure-specificity of RDA ene-2-carboxylic acid with formylbenzoic acid followed phenomena have previously been studied by us in the by a mild thermolysis of the pentacyclic intermediate mass spectra of stereoisomeric polycondensed hetero- afforded the novel 1,3-oxazino[2,3-a]indole-2,6-dione cycles using various ionization methods, such as elec- [1]. -
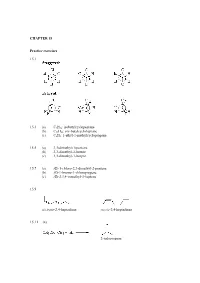
CHAPTER 15 Practice Exercises 15.1 15.3 (A) C9H18
CHAPTER 15 Practice exercises 15.1 15.3 (a) C9H18: isobutylcyclopentane (b) C11H22: sec-butylcycloheptane (c) C6H2: 1-ethyl-1-methylcyclopropane 15.5 (a) 3,3-dimethyl-1-pentene (b) 2,3-dimethyl-2-butene (c) 3,3-dimethyl-1-butyne 15.7 (a) (E)-1-chloro-2,3-dimethyl-2-pentene (b) (Z)-1-bromo-1-chloropropene (c) (E)-2,3,4-trimethyl-3-heptene 15.9 cis,trans-2,4-heptadiene cis,cis-2,4-heptadiene 15.11 (a) 2-iodopropane (b) 1-iodo-1-methylcyclohexane 15.13 CH3 CH3 + HI I Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step + CH3 HI CH3 I rate-determining step Step 2: Nucleophilic attack of the iodide anion on the 3˚ carbocation intermediate to give the product: CH + 3 CH3 I I 15.15 H2O CH3 CH3 + H3O OH Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step H + + CH3 HOH CH3 O H rate-determining H step Step 2: Nucleophilic attack of the water on the 3˚ carbocation intermediate to give the protonated alcohol: H H O+ H + CH3 O H CH3 Step 3: The protonated alcohol loses a proton to form the product: H + O H H CH3 + O + H3O CH3 H OH 15.17 (a) 2-phenyl-2-propanol (b) (E)-3,4-diphenyl-3-hexene (c) 3-methylbenzoic acid or m-methylbenzoic acid Review questions 15.1 A hydrocarbon is a compound composed only of hydrogen and carbon atoms. 15.3 In saturated hydrocarbons, each carbon is bonded to four other atoms, either hydrogen or carbon atoms. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each.