Linkage Mapping, QTL Analysis and Population Genomic Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Eucalypt Cooperative
Eucalypt Cooperative Report No.8 Realized gain and accuracy of breeding- value estimation for first- generation Eucalyptus nitens planted in New Zealand by Rafael Zas and Ruth McConnochie Realized gain and accuracy of breeding- value estimation for first- generation Eucalyptus nitens planted in New Zealand by Rafael Zas 1 and Ruth McConnochie 2 1Departamento de Producción Forestal, Centro de Investigacións Forestais e Ambientais de Lourizán, Apdo. 127, Pontevedra, 36080, Spain 2Scion-Ensis Genetics, Private Bag 3020, Rotorua, New Zealand Date: March 2007 Client: Eucalypt Cooperative Disclaimer: The opinions provided in the Report have been prepared for the Client and its specified purposes. Accordingly, any person other than the Client, uses the information in this report entirely at its own risk. The Report has been provided in good faith and on the basis that every endeavour has been made to be accurate and not misleading and to exercise reasonable care, skill and judgment in providing such opinions. Neither Ensis nor its parent organisations, CSIRO and Scion, or any of its employees, contractors, agents or other persons acting on its behalf or under its control accept any responsibility or liability in respect of any opinion provided in this Report by Ensis. 2 Eucalypt Cooperative REPORT No: 8 Table of Contents Summary 3 Introduction 4 Material and methods 5 The trials 5 Assessments 6 Statistical analysis 6 Results and Discussion 8 Conclusions 11 Acknowledgments 11 References 12 APPENDIX 1 13 APPENDIX 2 15 3 Eucalypt Cooperative REPORT No: 8 Realized gain and accuracy of breeding-value estimation for first-generation Eucalyptus nitens planted in New Zealand By Rafael Zas and Ruth McConnochie Summary This paper reports the results at age seven years of two Eucalyptus nitens genetic gain trials established in New Zealand with different seedlots of open- pollinated first-generation families grouped according to the parental breeding values (BV) for volume and wood density. -

RFPG Submission to Murrindindi Shire Council Re Tourist Development
Rubicon Forest Protection Group Submission to Murrindindi Shire Council on the Draft 2019-20 Budget In June 2016 the Rubicon Forest Protection Group made a presentation to the then Murrindindi Council on the fate awaiting the Rubicon State Forest if logging continued as planned. Our submission pointed to the amazing tourist potential of the area and how this was being ruined by the scale of logging on top of the vast area of ash forest killed in the 2009 fire. Distressingly, logging has continued largely unabated. The Royston Range, Middle Range and Rubicon Valley are now effectively logged out severely compromising their tourism potential, yet VicForests propose even more coupes in these areas. However the main focus of logging has shifted eastward to the relatively lightly logged Torbreck Range, to the eastern side of the Snobs Creek Valley and southward to Cambarville. The prospect of widespread logging of these extraordinarily biodiverse and beautiful areas is utterly appalling. We again now urge Council to do more to help protect what is left of the over-logged forests in our Shire, by developing and promoting their extraordinary tourist potential. This includes pressing the State Government to do more to promote and protect the forest, and help our Shire build a tourism future involving its wonderful natural assets. In terms of pressing the State Government, we ask that as part of the tourism and events strategy which Council is developing (but about which we know little) provision is made in the 2019-20 Budget to establish a Murrindindi Forest Tourism Development and Promotion Committee (MFTDPC). -

Walk-Issue02-1951.Pdf
Terms and Conditions of Use Copies of Walk magazine are made available under Creative Commons - Attribution Non-Commercial Share Alike copyright. Use of the magazine. You are free: • To Share-to copy, distribute and transmit the work • To Remix- to adapt the work Under the following conditions (unless you receive prior written authorisation from Melbourne Bushwalkers Inc.): • Attribution- You must attribute the work (but not in any way that suggests that Melbourne Bushwalkers Inc. endorses you or your use of the work). • Noncommercial -You may not use this work for commercial purposes. • Share Alike- If you alter, transform, or build upon this work, you may distribute the resulting work only under the same or similar license to this one. Disclaimer of Warranties and Limitations on Liability. Melbourne Bushwalkers Inc. makes no warranty as to the accuracy or completeness of any content of this work. Melbourne Bushwalkers Inc. disclaims any warranty for the content, and will not be liable for any damage or loss resulting from the use of any content. For Happy Hiking . CONSULT THE VJ(JTORIAN G01TERNMENT TOURIST BUREAU 272 COLLINS STREET, MELBOURNE 'PHONE F.0404 WALK THE JOURNAL OF THE MELBOURNE BUSHWALKERS No.2 1951 CONTENTS EDITORIAL .. ...................• . .. .. .. .. .. .. 3 FUJISAN .. ................. G. McKinnon . 4 ROAMING AROUND THE GORGES N. Richards . 8 THE ROCKING STONE SADDLE .. ]. Smith ...... ll "THE BUSHWALKERS" . Anon . ........ 12 SOME NOTES ON AN OLD SONG . F. Pitt . 13 CHARLES LESLIE GREENHILL - OBIT. .. .. .. .. .. .. 15 BUSHWALKING IN AUSTRALIA AND NEW ZEALAND ................ G. Bruere .. 17 THE MENAGERIE LION ........................ 21 A PLEA FOR POSTERITY . G. Coutts . 24 ON THE ART OF PHOTOGRAPHY . -

Tourist Development Proposals for the Rubicon State Forest
Tourist Development Proposals for the Rubicon State Forest 1. Upgrade Tweed Spur track and Cathedral Lane/Chitty Ridge Track to 2WD standard to create a wonderful scenic drive linking the Cathedral Range State Park to the Rubicon State Forest Tweed Spur track (yellow on map below) runs from Cooks Mill in the Cathedral Range State Park to Blue Range Road (mauve on the map) on the Cerberean Plateau and while officially 4WD is of a gentle grade and in fair condition. Chitty Ridge track (orange) also has a gentle grade and is nominally 2WD but its present condition makes it effectively 4WD. The eastern end of Cathedral lane (light blue), which is a shire road, is in very poor condition and is slow going in 4WD. If this sequence of tracks (apart from Blue Range Rd which is fine) was upgraded visitors to the Cathedral State Park would have the option of a wonderful forest drive with fabulous views (from Chitty Ridge Track). The geological history of the Cerberean Plateau (remnants of a massive extinct volcano) is fascinating and there are various road cuttings where descriptive signage could be installed explaining this history, as well as signs illustrating the logging and fire history of the various forest stands that are encountered. A walking track could also be constructed to the Little River Falls from Tweed Spur Road, as could a camping area on Blue Road Range adjacent to a lovely creek. Rubicon Forest Protection Group www.rubiconforest.org 2. Develop other self-guided driving tours on the Plateau highlighting historic, ecological and geologically significant spots At present DELWP produce several ‘Forest Notes’ brochures, one focused on the Rubicon Historic Area and one on the Snobs Creek area, but these are now rather dated. -

Forest Management Plan for the Central Highlands 1998
FOREST MANAGEMENT PLAN FOR THE CENTRAL HIGHLANDS Department of Natural Resources and Environment May 1998 ii Copyright © Department of Natural Resources and Environment 1998 Published by the Department of Natural Resources and Environment PO Box 500, East Melbourne, Victoria 3002 Australia http://www.nre.vic.gov.au This publication is copyright. Apart from any fair dealing for private study, research, criticism or review as permitted under the Copyright Act 1968, no part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, photocopying or otherwise, without prior permission of the copyright owner. Victoria. Department of Natural Resources and Environment Forest management plan for the Central Highlands. Bibliography. 1. Forest management - Environmental aspects - Victoria - Central Highlands Region. 2. Forest conservation - Victoria - Central Highlands Region. 3. Forest ecology - Victoria - Central Highlands Region. 4. Biological diversity conservation - Victoria - Central Highlands Region. I. Title. 333.751609945 General Disclaimer This publication may be of assistance to you but the State of Victoria and its employees do not guarantee that the publication is without flaw of any kind or is wholly appropriate for your particular purposes and therefore disclaim all liability for any error, loss or other consequences which may arise from you relying on any information in this publication. Printed by Gill Miller Press Pty. Ltd. ISBN 0 7311 3159 2 (online version) iii FOREWORD Extending from Mt Disappointment in the west to Lake Eildon and the Thomson Reservoir, the forests of the Central Highlands of Victoria contain major environmental, cultural and economic resources. -

Melbourne Area District 2 Review
LAND CONSERVATION COUNCIL MELBOURNE AREA DISTRICT 2 REVIEW FINAL RECOMMENDATIONS July 1994 This text is a facsimile of the former Land Conservation Council’s Melbourne Area District 2 Review Final Recommendations. It has been edited to incorporate Government decisions on the recommendations made by Orders in Council dated 5 September 1995 and 17 June 1997 and formal amendments. Subsequent changes may not have been incorporated. Where the Review refers back to the January 1977 Melbourne Area Final Recommendations, for completeness recommendation wording and Crown descriptions have been reproduced. Added text is shown underlined; deleted text is shown struck through. Annotations [in brackets] explain the origin of changes. 2 MEMBERS OF THE LAND CONSERVATION COUNCIL D.M. Calder, M.Sc., Ph.D., M.I.Biol. (Deputy Chairman) P.J. Dowd, B.Sc.(Eng.); Deputy Secretary, Resources Development, Department of Energy and Minerals M.D.A. Gregson, E.D., M.A., F. of Aus I.M.M.; Deputy Secretary Minerals, Department of Energy and Minerals R.L. Leivers Dip.Agr.Sc; B.Agr.Sc.(Hons); Acting Director, Catchment and Land Management, Department of Conservation and Natural Resources. R.D. Malcolmson, MBE., B.Sc., F.A.I.M., M.I.P.M.A., M.Inst.P., M.A.I.P. B. Nicholls, M.Ec., B.Ec., Hons. (1st Class), TPTC; Secretary, Department of Planning and Development. P. Price, B.Sc, Dip.Ed.; R.P. Rawson, Dip.For.(Cres.), B.Sc.F. Director, Forest Services, Department of Conservation and Natural Resources D. Robinson, B.Sc.(Hons.), Ph.D. D.S. Saunders, B.Agr.Sc.; Director, National Parks, Department of Conservation and Natural Resources P.G. -
Snobs Creek Falls
Snobs Creek Falls W ATERFALL S ERIES SNOBS CREEK FALLS CONTRIBUTORS State Library Victoria Murrindindi Library Services Shannon Carnes James Cowell Sandra Cumming Travis Easton: (Author of Melbourne Waterfalls - www.telp.com.au) Rod Falconer Ted & Val Hall Lawrence Hood Allan Layton Leisa Lees Kathie Maynes Ken Nash John & Maureen Norbury Kelly Petersen David & Debbie Hibbert # FACTSHEET 025 CONTENTS INTRODUCTION TIMELINE GALLERY NEWSPAPERS ESPLASH HISTORIC FACTSHEET I NTRODUCTION Name: Snobs Creek Falls Snobs Creek Falls was previously called Previously: Niagara Falls Yarram Falls and Niagara Falls, and was once referenced as the 'Great Falls of Location: 5.7 km from Snobs Creek Australia's Niagara'. At this time it was Drop: 110 metres (360 feet) considered to be Alexandra's most GPS: S37.30391 E145.88824 important and spectacular tourist attraction. The Upper Snobs Creek Falls This viewing position is no longer accessible In the late 1800s and early 1900s, the Snobs Creek Falls were known as 'Niagara Falls' and 'Alexandra Falls' and were considered the most important tourist attraction in the Alexandra district. It was also referred to as the 'Snobb's Creek Falls' in the Alexandra Standard and on at least one George Rose Postcard. At that time a walking track led from the camping area (located further downstream near where the highway is today) to the 'Lower' Falls. The track then continued to the 'Upper' Snobs Falls and then the Banyarmbite Cascades, located further upstream again. Page 4 HISTORIC FACTSHEET I NTRODUCTION The track was maintained by the Shire of Alexandra, included a system of stairs cut into the steep hillside, and gave access to the entire complex. -
Taungurung Clans Brochure
Consequences The Taungurung and other members of While travelling through Taungurung lands Many Taungurung people still live on Kulin Nation were deeply impacted by you will be aware of the following towns. their country and participate widely in the of Colonialism the dictates of the various government All these towns have a Taungurung origin: community as Cultural Heritage Advisors, assimilation and integration policies. Land Management Officers, artists and When Europeans first settled the region Benalla Today, the descendants of the Taungurung educationalists and are a ready source of in the early 1800s, the area was already Benalta = Big waterhole form a strong and vibrant community. knowledge concerning the Taungurung occupied by Taungurung people. From Descendants of five of the original clan Delatite people from the central areas of Victoria. that time, life for the Taungurung people groups meet regularly at Camp Jungai – Delotite, wife of Beeolite, clan head of the We are pleased to welcome you to our in central Victoria changed dramatically an ancestral ceremonial site. Yowung-Illam-Balluk clan country – to enjoy the landscapes, the flora and was severely disrupted by the early Murrindindi and fauna The Taungurung will continue establishment and expansion of European Elders assist with the instruction of Murrumdoorandi = Place of mists, to care for this country and welcome those settlement. Traditional society broke down younger generations in culture, history, mountain who share a similar respect. with the first settler’s arrival and soon and language and furthering of their after, Aboriginal mortality rates. soared as knowledge and appreciation of their Trawool For further information please a result of introduced diseases, denial of heritage as the rightful custodians of the Tarawil = Turkey contact: Taungurung lands in Central Victoria. -

Submission from RFPG on Timber Release Plan (TRP)
Submission from RFPG on Timber Release Plan (TRP) Proposed Changes, August 2020 Since its inception in 2015, the Rubicon Forest Protection Group (RFPG) has been drawing attention to the total unsustainability of logging in the Rubicon State Forest. Now, in the wake of the recent bushfires in northeast Victoria and East Gippsland, an already dire situation has become worse. With 4 massive bushfires this century leaving little of eastern Victoria untouched, we may now be only one fire away from mass extinction of forest dwelling animals. Under these circumstances, native forest logging needs to wind down well before 2024 and cease far sooner than 2030 if the principles and a number of the mandatory provisions of the 2014 Code of Practice for Timber Production (the Code) are to be met. Our comments on the latest TRP change proposals focus on their failure to meet the requirements of the Code principally in the Rubicon State Forest, but also other State Forests in the Central FMA including the Mt Disappointment, Tallarook, Kinglake, Toolangi, Black Range, Marysville and Big River State Forests. We cite breaches that call into question not only VicForests’ planning failures, but the failure of the VicForests Board to properly discharge its obligations. While VicForests has largely ignored our past submissions and made few substantive adjustments to its plans, Minister Symes’ confirmation that current timber harvesting is indeed unsustainable requires that our criticisms of the latest proposals be properly evaluated. If not, VicForests needs to be ready to face further legal action, including against the Board since it is responsible for managing VicForests’ affairs. -
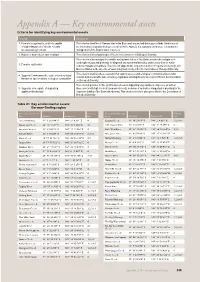
Key Environmental Assets Criteria for Identifying Key Environmental Assets
Appendix A — Key environmental assets Criteria for identifying key environmental assets Criterion Explanation 1. Formally recognised in, and/or is capable This criterion identifies all Ramsar sites in the Basin and any wetland that supports birds listed in any of of supporting species listed in, relevant the international migratory bird agreements to which Australia is a signatory and that are relevant to the international agreements management of the Basin’s water resources. 2. Natural or near-natural, rare or unique This criterion is developed to give effect to the Convention on Biological Diversity. This criterion acknowledges the variable and dynamic nature of the Basin, and includes refugia such as drought refuges and pathways for dispersal and ephemeral breeding, and nursery sites for water- 3. Provides vital habitat dependent plants and animals. This criterion supports the long-term retention of regional biodiversity, and thus contributes to the objective of conserving biodiversity under the Convention on Biological Diversity. This criterion identifies the ecosystems that support species and ecological communities listed under 4. Supports Commonwealth-, state- or territory-listed relevant Commonwealth, state or territory legislation as being threatened. It gives effect to the Convention threatened species and/or ecological communities on Biological Diversity. This criterion provides for the identification of assets supporting large numbers of species, as well as 5. Supports, or is capable of supporting, those assets with high levels of taxonomic diversity. Inclusion of such sites is important in providing for the significant biodiversity long-term viability of the Basin’s biodiversity. This criterion therefore also gives effect to the Convention on Biological Diversity. -

Part 1 Introduction
1 Part 1 Introduction Fishing is one of the most popular recreational Victorian recreational fishers for recreational pursuits in Victoria, with more than 800,000 fisheries in the upper Goulburn River Victorians participating in fishing at least once catchment. a year (Tourism Victoria 1995). The recreational fishing sector (including support industries) In doing so it must: generates approximately 27,000 associated jobs ● ensure that the use and management of the and is estimated to contribute approximately fishery resource is consistent with the $1.265 billion per annum to the Victorian principles of ecological sustainability; economy (Unkles 1997). ● ensure that the management of the fishery is consistent with other conservation and A fisheries management plan is a public natural resource management aims of the document which clearly outlines future Department of Natural Resources and management directions and arrangements for Environment (DNRE) and other agencies recreational and/or commercial fishing for (such as Goulburn-Murray Water, Goulburn individual water-bodies or species within a Broken Catchment Management defined area. Authority); and As the Government agency responsible for ● develop guidelines for the resolution of fisheries, the role of Fisheries Victoria is to be issues between user groups. a lead advocate for fish habitat protection in The development of the GERFMP involves public waters and to manage recreational, direct participation by Victorian recreational commercial and traditional fisheries and fishers. It operates in an inclusive manner to commercial aquaculture in Victorian waters. ensure that the wider stakeholder groups are In doing so, Fisheries Victoria recognises and informed of progress and have every supports the need for protection of species opportunity to have input into the GERFMP. -

Goulburn River Reach Report: Eildon Dam to Killingworth
Goulburn reach report, Constraints Management Strategy Eildon Dam to Killingworth At a glance The flow footprints constraints work is investigating are 12,000–20,000 megalitres/day (ML/d) between Eildon Dam and the Killingworth gauge on the Goulburn River, including the flow contributions of tributaries. Extended duration releases from Lake Eildon and releases on top of high tributary flows are not being investigated. Flow footprints higher than 20,000 ML/d are not being investigated, which are large events like 2010 which reached over 40,000 ML/d at Trawool. The overbank flows that constraints work is looking at occurred more frequently in the past. Feedback from local councils and landholders was that the 12,000 ML/d flow footprint map looked about right and would not be expected to cause too many issues. However, the 15,000 ML/d and 20,000 ML/d flow maps underestimate the flow footprint. Initial feedback from landholders suggests that up to a maximum 12,000 ML/d flow footprint around Molesworth and a 15,000 ML/d flow footprint elsewhere in the subreach may be a tolerable level of inconvenience flooding if suitable mitigation measures are put in place. Flows of up to 12–15,000 ML/d could be created by releases from Eildon with no tributary inflows, tributary flows on their own, or a mix of tributary flows and Eildon releases. Reach characteristics This subreach flows from Eildon Dam to the Killingworth gauge (405329) on the Goulburn River (Figure 15). Water in this stretch of river comes from releases from Eildon Dam and inflows from several unregulated tributaries.